Abstract
A simple, sensitive and selective cloud point extraction procedure is described for the preconcentration and atomic absorption spectrometric determination of Zn2+ and Cd2+ ions in water and biological samples, after complexation with 3,3',3",3'"-tetraindolyl (terephthaloyl) dimethane (TTDM) in basic medium, using Triton X-114 as nonionic surfactant. Detection limits of 3.0 and 2.0 µg L-1 and quantification limits 10.0 and 7.0 µg L-1were obtained for Zn2+ and Cd2+ ions, respectively. Relative standard deviation was 2.9 and 3.3, and enrichment factors 23.9 and 25.6, for Zn2+ and Cd2+ ions, respectively. The method enabled determination of low levels of Zn2+ and Cd2+ ions in urine, blood serum and water samples.
Zn2+ and Cd2+ ions; TTDM; flame atomic absorption
ARTIGO
Application of cloud point preconcentration and flame atomic absorption spectrometry for the determination of cadmium and zinc ions in urine, blood serum and water samples
Ardeshir ShokrollahiI,* * e-mail: ashokrollahi@mail.yu.ac.ir ; Somayeh JoybarI; Hoda Elias HaghighiII; Khodabakhsh NiknamIII; Ebrahim NiknamIII
IDepartment of Chemistry, Yasouj University 75918-74831, Yasouj, Iran
IIChemistry Department, Islamic Azad University of Firouzabad, Firouzabad 74715-117, Iran
IIIChemistry Department, Persian Gulf University, Bushehr, Iran
ABSTRACT
A simple, sensitive and selective cloud point extraction procedure is described for the preconcentration and atomic absorption spectrometric determination of Zn2+ and Cd2+ ions in water and biological samples, after complexation with 3,3',3",3'"-tetraindolyl (terephthaloyl) dimethane (TTDM) in basic medium, using Triton X-114 as nonionic surfactant. Detection limits of 3.0 and 2.0 µg L-1 and quantification limits 10.0 and 7.0 µg L-1were obtained for Zn2+ and Cd2+ ions, respectively. Relative standard deviation was 2.9 and 3.3, and enrichment factors 23.9 and 25.6, for Zn2+ and Cd2+ ions, respectively. The method enabled determination of low levels of Zn2+ and Cd2+ ions in urine, blood serum and water samples.
Keywords: Zn2+ and Cd2+ ions; TTDM; flame atomic absorption.
INTRODUCTION
Zinc is one of the least common elements and has been estimated to make up 0.0005-0.02% of the earth's crust. It is an essential element in the growth of many kinds of organisms, both plants and animals. Zinc is present in all body tissues and fluids1 and is an essential component of a large number (>300) of enzymes participating in the synthesis and degradation of carbohydrates, lipids, proteins, and nucleic acids, as well as in the metabolism of other micronutrients. This metal is a cofactor of several enzyme systems and constitutes the active center of carbonic anhydrase. The insulin compound is a zinc-containing protein while zinc is also present in most foods, particularly those high in protein.2 Zinc stabilizes the molecular structure of cellular components and membranes and in this way contributes to the maintenance of cell and organ integrity. Furthermore, zinc plays an essential role in polynucleotide transcription and thus in the process of genetic expression. Its involvement in such fundamental activities probably accounts for the essential nature of zinc for all life forms. Moreover, zinc plays a central role in the immune system, affecting a number of cellular and humeral immunity aspects.3 Zinc deficiency in the human diet has been found to retard growth and maturity and also to cause anemia. On the other hand, zinc is a man-made environmental pollutant. The concentration of zinc in unpolluted natural water is low.
The main uses of cadmium include nickel-cadmium batteries, invented by Thomas A. Edson in the early part of the 20th century, and in cadmium coatings for the anti-corrosion protection of steel.4 Our environment contains countless sources of cadmium and in the modern world this element is ubiquitous in food, water and air. The greatest human cadmium exposure comes from tobacco smoke and food consumption, most of which arises from the uptake of cadmium by plants from fertilizers, sewage sludge, manure and atmospheric deposition.5 Cadmium is not only regarded as essential to human life, but is now known to be extremely toxic6,7 and accumulates in humans mainly in the kidneys.4 Prolonged intake, even of tiny amounts, leads to severe dysfunction of the kidneys. Cadmium is widely found in society, either in useful products or in controlled wastes.
The sensitivity of analytical techniques is often insufficient for its determination. Several methods have been developed to separate and determine Zn2+ or Cd2+ ions. Traditionally, organic solvents8-12 have been used to extract Zn2+ or Cd2+ ions. However, these solvents are often classified as having carcinogenic toxicity and being environmental pollutants. In general, colorimetry is the conventional method commonly used in many fields. Nevertheless, almost none of the color reagents are specific for determining zinc or cadmium in complex matrices although their determination has been developed.13-16 In addition, although some of these methods13,15 number among the most accurate, they pose some associated risks for both the analysts and the environment. Therefore, the determination of zinc or cadmium concentration in environmental and biological settings by atomic absorption spectroscopy (AAS) represents an effective method. Nevertheless, this method requires a relatively large amount of sample in comparison with coupled techniques with AAS.
Aqueous solutions of many non-ionic surfactant micellar systems become turbid over a narrow temperature range, when the experimental conditions (i.e., temperature or pressure, addition of salt or other additives, etc.) are changed. This temperature is called the cloud point temperature. Above the cloud point, the aqueous surfactant micellar solution separates into a concentrated phase containing most of the surfactant (termed surfactant-rich phase) and a dilute aqueous phase containing low concentration of surfactant corresponding to critical micellar concentration (cmc). Any components originally present that bind to the micellar aggregate in solution can thus be extracted from the original solution and concentrated in the small volume element of the surfactant-rich phase (usually less than 0.5 mL).
In contrast to traditional liquid-liquid extraction (LLE), cloud point extraction (CPE) has little or no pollution effect. There is another advantage of the CPE method, in as far as a concentration effect can been expected as a function of the small volume of the separated surfactant phase.17 Thus, there is a growing tendency to use surfactants in trace analysis of organic18-21 and inorganic materials.22-32
However, despite the critical importance of Zn2+ and Cd2+ monitoring in many industrial, environmental, clinical and pharmaceutical samples, only a few cases of the use of CPE for the separation and preconcentration of Zn2+ and Cd2+ have been reported in the literature.33-36
In this work, a cloud-point preconcentration procedure was introduced for determination of Zn2+ and Cd2+ ions, after the formation of complex with 3,3',3",3'"-tetraindolyl (terephthaloyl) dimethane (TTDM). The lipophilic TTDM-Zn2+ and Cd2+ complexes, completely extracted from aqueous solution to the concentrated micellar medium, and the analytes were ultimately analyzed by flame atomic absorption spectrometry.
EXPERIMENTAL
Reagents
All solutions were prepared with deionized water. Analytical-grade methanol, acids, and other chemicals used in this study were obtained from Merck. A 1.0% (w/v) Triton X-114 from E. Merck (Darmstadt, Germany) was prepared by dissolving 1.0 g of Triton X-114 in a 100 mL volumetric flask with stirring. All chemicals such as nitrate of Zn(II) and Cd(II) and other cations were of analytical grade purchased from Merck. The 3,3',3",3'"-tetraindolyl (terephthaloyl) dimethane (TTDM) was synthesized, purified and characterized according to the literature.37,38
Synthesis of TTDM
To synthesize TTDM (Figure 1), a mixture of indole (4 mmol, 0.468 g), terephthaldehyde (1 mmol, 0.134 g), and Al(HSO4)3 (2 mmol, 0.636 g) in dimethyl sulfoxide (5 mL) was heated in an oil bath at 120 ºC. The reaction mixture was filtered after 1 h. The residue was washed with ethanol (2 × 4 mL). water (40 mL) was then added to the filtrate, and the precipitate filtered. The product was purified by column chromatography on silica gel, using eluent: ethyl acetate/n-hexane (10:90)] to give pure tetra(indolyl)methane 0.492 g, 87% yield. Purification and characterization was achieved according to our previous report. 38
Instrumentation
A Perkin-Elmer model 97 AAnalyst 300 (Shelton, CT, USA) atomic absorption spectrometer equipped with deuterium background correction, Zn and Cd hollow-cathode lamps as the radiation source, was used for absorbance measurements at wavelengths of 213.9 and 228.8 nm, respectively. The instrumental parameters were adjusted according to the manufacturer's recommendations. A UV-Vis spectrophotometer Jasco model V-570 (Japan) was used for spectrophotometric titrations. An A30 E 148 centrifuge was used to accelerate the phase separation process. A Metrohm 744 pH meter, furnished with a combined glass-saturated calomel electrode, was used for pH measurements.
Spectrophotometric titrations
Standard stock solutions of TTDM (103 M) and the metal ions (3.0 × 103 M) were prepared by dissolving appropriate and exactly weighed (accuracy of ± 0.0001 g) amounts of pure solid compounds in pre-calibrated 25.0 mL volumetric flasks, and diluting to the mark with MeOH. Working solutions were prepared by appropriate dilution of the stock solutions. Titration of 2.6 mL of TTDM 2.3 × 104 M solution was carried out by the addition of micro-liter amounts of a concentrated standard solution of the metal ion (1.0-2.5 × 103 M) using a pre-calibrated micro-syringe, at 25.0 ºC, with the spectrum subsequently recorded.
General procedure
A typical cloud point experiment required the following steps: an aliquot of 15 mL of a solution containing 66.7 µg L-1 Zn2+ and 133.3 µg L-1 Cd2+ ions, 0.08% (w/v) Triton X-114 and TTDM (0.08 mM) was adjusted to pH 10 by adding NaOH. The mixture was shaken for 1 min and left to stand in a thermostated bath at 65 ºC, for 25 min. Separation of the phases was achieved by centrifugation at 3500 rpm for 15 min. The whole system was cooled in an ice bath for 5 min to allow the surfactant rich phase to regain its viscosity. In this way, the bulk aqueous phase was easily decanted. The remaining micellar phase was dissolved to 0.5 mL in 1.0 M HNO3 in methanol and the Zn2+ and Cd2+ ions content was readily evaluated by flame atomic absorption spectroscopy (FAAS).
Application of real samples
Water samples
Analysis of water samples for determination of analyte contents was performed as follows: about 500 mL of sample was passed through a sintered glass funnel and 1 mL of concentrated HNO3 was added to eliminate and decompose micro-organisms and acidify the medium. The water samples were stored in polyethylene bottles. The procedure given in the general procedure section was then applied for analysis.
Blood serum samples
A 5-mL portion of the clear supernatant serum obtained from 2 samples from males or females aged 28 and 32 years was pipetted off and treated with 5.0 mL of 1.2 M trichloroacetic acid. The mixture was centrifuged at 3000 rpm, the deproteinized supernatant placed in a 50-mL beaker, and the resulting solution neutralized with 2.0 M NaOH until a pH value of ca10 was reached.23 Aliquots (7-mL) of the samples were then analyzed by the proposed method.
Urine samples
A 10 mL portion of a urine sample (or a spiked urine sample) was treated with 10 mL of concentrated HNO3 (63%) and an HClO4 (70%) mixture of 2:1 in a 50 mL beaker covered with a watch glass. The content of the beaker was heated on a hot plate (100 ºC 15 min, 150 ºC 10 min). The watch glass was removed and the acid evaporated to dryness at 150 ºC. HClO4 (3 mL) was added to the resulting white residue and the mixture was heated at 160 ºC to dryness. All heating was carried out under a hood while taking the necessary precautions. Five milliliters of 1 M H2SO4 was added, the mixture heated at 150 ºC for 1 min, and the volume made up to the mark in a 50 mL volumetric flask. Aliquots (7 mL) of the resulting clear solution were analyzed according to the described procedure.39
RESULTS AND DISCUSSION
The aim of this work was to develop a simple, sensitive and available method for the preconcentration and determination of trace amounts of Zn2+ and Cd2+ ions in various real samples using flame atomic absorption spectrometry coupled with CPE. In this regard, the influence of various effective parameters including, pH, surfactant and TTDM concentrations, heating time and temperature, centrifuge time and rate, as well as the effect of electrolyte on absorbance, were optimized.
The complexation study yields important information about the interaction between the ligand and metal ions. Recently, we have used the spectrophotometric method for this purpose25 before using TTDM for the CPE of the metal ions.
Spectrophotometric investigation
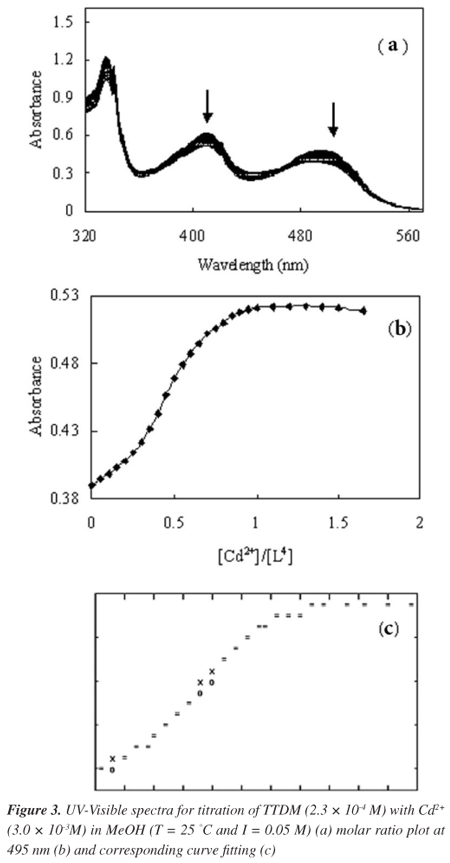
The spectra of TTDM with the addition of Zn2+ or Cd2+ ions, corresponding mole ratio plot and curve fitting at wavelengths of 498 or 495 nm, are depicted in Figures 2 and 3, respectively. Complexation occurs through binding of Zn2+ or Cd2+ ions to nitrogen atoms, leading to the ML, or ML, ML2 models. Formation constants and stoichiometries of complexes were calculated using the Kinfit program25,40-43 and results are shown in Table 1.
Effect of pH
The formation of the metalchelate and its chemical stability are two important factors influencing CPE. The pH plays a unique role in metalchelate formation and subsequent extraction, proving to be the main parameter for CPE. Extraction yield depends on the pH at which complex formation is carried out. Thus, a set of similar experiments in the pH range 2.0-11.0 was conducted according to the described procedure described in the Experimental section with respective results illustrated in Figure 4. The maximum sensitivity by CPE was obtained at pH 10. In more acidic solutions, deterioration of the signal occurs due to protonation of TTDM, while at pH > 10, the signal decreases and recovery is reduced due to precipitation of Zn2+ and Cd2+ ions in the form of hydroxides or ternary complexes. Consequently, pH = 10 was selected for the subsequent studies.
Effect of TTDM concentration
The absorbance of extracted metal ions as a function of the concentration of TTDM is shown in Figure 5. As can be seen, increasing TTDM concentration up to a 0.08 mM causes increasing absorbance. Thus, a TTDM concentration of 0.08 mM was chosen to optimize other variables. At lower than 0.08 mM of TTDM, insufficient concentration leads to incomplete complexation. A possible explanation for the decrease in absorption with higher concentrations may be attributed to the formation of charged complexes with excess TTDM in the medium or with remaining excess TTDM in aqueous solution, competing with surfactant-rich phase for formation of complex with metal ions in aqueous phase.
Effect of Triton X-114 concentration
The type and concentration of surfactant are important factors in cloud point extraction. Triton X-114 was chosen as a suitable surfactant due to its physicochemical characteristics, low cloud point temperature, commercial availability, relatively low price, low toxicity, high density in the surfactant-rich phase and because it facilitates phase separation. The variation in absorbance of extracted Zn2+ and Cd2+ ions within the Triton X-114 concentration range of 6.6 ' 10-3- 0.16% (w/v) was examined and results shown in Figure 6. The results show that quantitative extraction was obtained with an optimum Triton X-114 concentration of 0.08% (w/v), at which the highest absorbance for extracted Zn2+ and Cd2+ ions was obtained. For concentrations lower than 0.08 % (w/v), the preconcentration efficiency of the formed complexes was very low, since the assemblies at low concentration were probably inadequate to preconcentrate trace amounts of Zn2+ and Cd2+ ions. The decreasing of absorbance at a concentration higher than 0.08% (w/v) is due to the remaining of some part of Triton X-114 and TTDM in aqueous solution as this phase can compete with surfactant-rich phase to draw analyte ions.
Effect of ionic strength
It is known that ionic strength of the solution is one of the effective factors in cloud point extraction. In this work, the effect of KNO3 salt as an electrolyte in the range 0.01-0.5 mol L1 on the process was investigated. It is observed that increasing concentration of salt to 0.1 M exerts no effect on the process, but higher concentrations caused a decrease in absorbance of extracted surfactant-rich phase. This effect might be explained by the additional surface charge when the salt concentration is very high.
Effect of temperature and equilibrium time on CPE
It was desirable to employ the shortest equilibration time and the lowest possible equilibration temperature as a compromise between completion of extraction and separation of phases. The dependence of extraction efficiency upon equilibration temperature and time above the cloud point in the range 30-70 ºC and 5-30 min were thoroughly optimized, respectively. Holding the sample solutions for 25 min at 65 ºC was found to be satisfactory to achieve a small volume of the surfactant-rich phase, quantitative extraction and experimental convenience.
Effect of centrifuge time and rate
Preconcentrating trace amounts of Zn2+ and Cd2+ ions with great efficiency in a short time is required. Therefore, CPE on a set of experiments using 15 mL samples under optimum conditions by heating to 65 ºC and centrifuging at various rates and times and subsequent cooling in 5 min was carried out. Centrifugation at 3500 rpm for 15 min separated the two phases completely. The enrichment phase did not separate completely at lower rates and centrifugation times.
Calibration, precision and detection limits
Calibration graphs were obtained by preconcentration of 15 mL of several solutions under optimum conditions, containing various concentrations of each analyte, with the process performed according to the General procedure section where the absorbance of the final solution was measured by FAAS. A linear relationship between the measured absorbance and concentration of Zn2+ or Cd2+ ions in the range 0.007-0.330 µg mL-1 was obtained. Table 2 shows the calibration parameters for the proposed CPE method including the linear ranges, the relative standard deviation obtained for two analyte samples subjected to the complete procedure, volume ratio and the limit of detection. Also, a calibration graph was obtained without preconcentration in order to calculate the enrichment factor. The enrichment factor was calculated as the ratio of the slopes of the calibration curve with and without preconcentration, and factors obtained for Zn2+ and Cd2+ ions were 23.89 and 25.60, respectively.
Interference study
With a view to obtaining high selectivity by the FAAS method, the interferences were studied. These interferences constitute special ions which are related to the preconcentration step, i.e. those that may react with TTDM and decrease the preconcentration and extraction efficiency (Table 3). The study of possible interferences was conducted for Li+ , K+ , Na+, Ni2+, Pb2+, Co2+, Ba2+, Ca2+, Mg2+, Cu2+, Mn2+, Fe3+, Cr3+, Al3+ , NO3, SO42-, ClO4, Cl- and Cd2+ or Zn2+ ions under the experimental conditions (at a fixed concentration of 66.7 or 133.3 µg L-1 of Zn2+ or Cd2+, respectively). There was no significant interference at a 1:1000 ratio of Zn2+ or Cd2+ with Li+, K+, Na+, Ni2+, Pb2+, Co2+, Ba2+, Ca2+, Mg2+, Cr3+ , NO3, ClO4, Cl- and SO42- ions, and also for the same condition at a ratio of 1:500 Zn2+ or Cd2+ with Cu2+, Mn2+, Fe3+, Al3+ , and Cd2+ or Zn2+ ions.
Real samples evaluation of method
The proposed method was applied for the determination of Zn2+ and Cd2+ ions by the standard addition technique in two blood serum, one urine and four different water samples including well water, spring water, tap water and mineral drinking water (Table 4). The results of two blood serum samples as mentioned in the Experimental section are summarized in Table 5. Notably, the determination of Zn2+ and Cd2+ in the blood serum samples is remarkably accurate by the proposed method. The results of the determination of Zn2+ and Cd2+ in the fresh urine sample as mentioned in the Experimental section are listed in Table 5.
CONCLUSION
The proposed cloud point extraction procedure using TTDM as a stable and fairly selective complexing agent offers a simple, rapid, inexpensive and environmentally benign method to preconcentrate and separate Zn2+ and Cd2+ in aqueous solutions. This method has a very low limit of detection (LOQ), good relative standard deviation (RSD) and was applied to determine trace amounts of Zn2+ and Cd2+ ions in various real samples. In a full comparison of the results presented in this paper with those using similar methods reported in literature1,7,8,13,43,44 (Table 6) revealed that, on key measurements, the proposed method proved comparable or superior to the reported methods.
Recebido em 27/3/12; aceito em 20/10/12; publicado na web em 21/2/13
- 1. Dallali, N.; Zahedi, M. M.; Yamini, Y.; Scientia Iranica 2007, 14, 291.
- 2. Kolachi, N. F.; Kazi, T. G.; Khan, S.; Wadhwa, S. K.; Baig, J. A.; Afridi, H .I.; Shah, A. Q.; Shah, F.; Food Chem. Toxicol. 2011, 49, 2548.
- 3. Shankar, A. H.; Prasad, A. S.; A.J.C.N 1998, 68, 447.
- 4. Greenwood, N. N.; Earnshaw, A.; Chemistry of the Elements, Pergamon Press, Oxford, 1989.
- 5. Davis, A. C.; Wu, P.; Zhang, X.; Hou, X.; Jones, B. T.; Appl. Spectrosc. Rev. 2006, 41, 35.
- 6. Cotton, F. A.; Wilkinson, G.; Murillo, C. A.; Bochmann, M.; Advanced Inorganic Chemistry, 6th ed., John Wiley & Sons Inc.: New York, 1999.
- 7. Ojeda, C. B.; Rojas, F. S.; Pavón, J. M.; Am. J. Anal. Chem. 2010, 1, 127.
- 8. Kilinc, E.; Cetin, A.; Togrul, M.; Hosgoren, H.; Anal. Sci. 2008, 24, 763.
- 9. Diantouba, B. A.; Guiguemde, I.; Tayeb, A.; Goetzgrandmont, G. J.; Brunette, J. P.; Ion Exch. Solvent Extr. 1994, 12, 325.
- 10. Sekine, T.; Kobelova, I.; Noro, J.; Nguyen, T. K. D.; Anal. Sci. 1994, 10,743.
- 11. Reddy, P. C.; Rangamannar, B.; J. Radioanal. Nucl. Chem. Lett. 1995, 200, 181.
- 12. Hammond, J. L-Ill.; Lee, Y.; Noble, C. O.; Beck, J. N.; Proffitt, C. E.; Sneddon, J.; Talanta 1998 ,47, 261.
- 13. Ghaedi, M.; Shokrollahi, A.; Niknam, K.; Niknam, E.; Najibi, A.; Soylak, M.; J Hazard. Mater. 2009, 168, 1022.
- 14. Xiang, G.; Wen, S.; Wu, X.; Jiang, X.; He, L.; Liu, Y.; Food Chem. 2012, 132, 532.
- 15. Makino, T.; Clin. Chim. Acta 1991, 197, 209.
- 16. Kaplan, A.; Jack, R.; Opheim, K. E.; Toivola, B.; Lyon, A. W.; Clinical chemistry interpretation and techniques; 4th ed., Williams & Wilkins: Malvern, 1995, p. 348.
- 17. Hinze, W. L.; Pramauro, E.; Crit. Rev. Anal. Chem. 1993, 24, 133.
- 18. Casero, I.; Sicilia, D.; Rubio, S.; Perez-Bendito, D.; Anal. Chem. 1999, 71, 4519.
- 19. Sicilia, D.; Rubio, S.; Perez-Bendito, D.; Maniasso, N.; Zagatto, E. A. G.; Anal. Chim. Acta 1999, 392, 29.
- 20. Pinto, C. G.; Perez-Pavon, J. L.; Moreno-Cordero, B.; Anal. Chem. 1994, 66, 874.
- 21. Du, M.; Wu, W.; Ercal, N.; Ma, Y.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2004, 803, 321.
- 22. Ghaedi, M.; Shokrollahi, A.; Mehrnoosh, R.; Hossaini, O.; Soylak, M.; Cent. Eur. J. Chem. 2008, 6, 488.
- 23. Shokrollahi, A.; Shamsipur, M.; Jalali, F.; Nomani, H.; Cent. Eur. J. Chem. 2009, 7, 938.
- 24. Shokrollahi, A.; Ghaedi, M.; Rajabi, H. R.; Ann. Chim. (Rome) 2007, 97, 823.
- 25. Shokrolahi, A.; Ghaedi, M.; Shabani, R.; Montazerozohori, M.; Chehreh, F.; Soylak, M.;Alipour, S.; Food Chem. Toxicol. 2010, 48, 482.
- 26. Ghaedi, M.; Daneshfar, A.; Shokrollahi, A.; Ghaedi, H.; Arvin Pili, F.; Ann. Chim. (Rome) 2007, 97, 971.
- 27. Shokrollahi, A.; Ghaedi, M.; Niband, M. S.; Rajabi, H. R.; J. Hazard. Mater. 2008, 151, 642.
- 28. Shokrollahi, A.; Ghaedi, M.; Hossaini, O.; Khanjari, N.; Soylak, M.; J. Hazard. Mater. 2008, 160, 435.
- 29. Shokrollahi, A.; Ghaedi, A.; Gharaghani, S.; Fathi, M. R.; Soylak, M.; Quim. Nova 2007, 31, 70.
- 30. Manzoori, J. L.; Bavili-Tabrizi, A.; Microchem. J. 2002, 72, 1.
- 31. Abdallah, A. M.; Kabil, M. A.; Akl, M. A.; Ismael, D. S.; J. Iran. Chem. Soc. 2004, 1, 79.
- 32. Ghazy, S. E.; El-Shazly, R. M.; El-Shahawi, M. S.; El-Hazmi, G. A. A.; El- Asmy, A. A.; J. Iran. Chem. Soc. 2006, 3, 140.
- 33. Ghaedi, M.; Shokrollahi, A.; Niknam, K.; Niknam, E.; Soylak, M.; Cent. Eur. J. Chem. 2009, 7, 148.
- 34. Ghaedi, M.; Shokrollahi, A.; Niknam, K.; Soylak, M.; Sep. Sci. Technol. 2009, 44, 773.
- 35. Chen, J.; Teo, K. C.; Anal. Chim. Acta 2003, 450, 215.
- 36. Kulichenko, S. A.; Doroshchuk, V. A. J.; Anal. Chem. 2003, 58, 524.
- 37. Niknam, K.; Zolfigol, M. A.; Sadabadi, T.; Nejati, A.; J. Iran. Chem. Soc. 2006, 3, 318.
- 38. Zolfigol, M. A.; Salehi, P.; Shiri, M.; Sayadi, A.; Abdoli, A.; Keypour, H.; Rezaeivala, M.; Niknam, K.; Kolvari, E.; Mol. Divers. 2008, 12, 203.
- 39. Shemirani, F.; Baghdadi, M.; Ramezani, M.; Jamali, M. R.; Anal. Chim. Acta 2005, 534, 163.
- 40. Dye, J. L.; Nicely, V. A.; J. Chem. Edu1971, 48, 443.
- 41. Shokrollahi, A.; Ghaedi, M.; Montazerzohori, M.; Kianfar, A. H.; Ghaedi, H.; Khanjari, N.; Noshadi, S.; Joybar, S.; E-J chem. 2011, 8, 495.
- 42. Shokrollahi, A.; Ghaedi, M.; Alipour, S.; Kianfar, A. H.; Eur. J. Chem. 2011, 2, 324.
- 43. Silva, E. L.; Roldan, P. dos S.; Gin, M. F.; J. Hazard. Mater. 2009, 171, 1133.
- 44. Hassanien, M. M.; Hassan, A. M.; Mortada, W. I.; El-Asmy, A.A.; Am. J. Anal. Chem. 2011, 2, 697.
Publication Dates
-
Publication in this collection
11 Apr 2013 -
Date of issue
2013
History
-
Received
27 Mar 2012 -
Accepted
20 Oct 2012