Resumos
O receptor de sulfoniluréia (SUR1) é uma subunidade dos canais de potássio ATP-dependentes expressos nas células beta pancreáticas, O papel deste receptor nos mecanismos de secreção da insulina foi bem demonstrado após a descrição de que mutações no seu gene codificador são responsáveis pela forma neonatal de hiperinsulinismo. O possível envolvimento de variantes deste gene na predisposição genética ao diabetes mellitus tipo 2 também tem sido estudado. Nesta revisão, discutimos os dados da literatura que abordam o envolvimento de alterações genéticas do SUR1 em patologias como o diabetes tipo 2, assim como nos mecanismos de secreção da insulina.
Receptor de sulfoniluréia; Gene SUR1; Diabetes mellitus; Genética
The sulfonylurea receptor is a subunit of the ATP-sensitive potassium channel, which is expressed in the pancreatic beta cell. The central role of this receptor in glucose-induced insulin secretion was confirmed by description that mutations in this gene might result in hyperinsulinemic hypoglycemia of infancy. The possible role of SUR1 gene variants in the genetic susceptibility for type 2 diabetes mellitus has been studied. In this review, we discuss the results concerning the genetic variations in SUR1 gene with diseases as type 2 diabetes mellitus and also with the mechanisms of insulin secretion.
Sulfonylurea receptor; SUR1 gene; Diabetes mellitus; Genetic
revisão
Patologia Molecular do Receptor de Sulfoniluréia (SUR1)
André F. Reis
Gilberto Velho
Disciplina de Endocrinologia,
Departamento de Medicina,
Universidade Federal de São
Paulo - Escola Paulista de
Medicina (UNIFESP/PEM),
São Paulo, SP, Brasil e
INSERM U342, Institut
Cochin de Génétique
Moléculaire (ICGM), Paris,
França
Recebido em 08/08/2000
Aceito em 15/08/2000
RESUMO
O receptor de sulfoniluréia (SUR1) é uma subunidade dos canais de potássio ATP-dependentes expressos nas células beta pancreáticas, O papel deste receptor nos mecanismos de secreção da insulina foi bem demonstrado após a descrição de que mutações no seu gene codificador são responsáveis pela forma neonatal de hiperinsulinismo. O possível envolvimento de variantes deste gene na predisposição genética ao diabetes mellitus tipo 2 também tem sido estudado. Nesta revisão, discutimos os dados da literatura que abordam o envolvimento de alterações genéticas do SUR1 em patologias como o diabetes tipo 2, assim como nos mecanismos de secreção da insulina. (Arq Bras Endocrinol Metab 2000;44/5: 382-389)
Unitermos: Receptor de sulfoniluréia; Gene SUR1; Diabetes mellitus; Genética
ABSTRACT
The sulfonylurea receptor is a subunit of the ATP-sensitive potassium channel, which is expressed in the pancreatic beta cell. The central role of this receptor in glucose-induced insulin secretion was confirmed by description that mutations in this gene might result in hyperinsulinemic hypoglycemia of infancy. The possible role of SUR1 gene variants in the genetic susceptibility for type 2 diabetes mellitus has been studied. In this review, we discuss the results concerning the genetic variations in SUR1 gene with diseases as type 2 diabetes mellitus and also with the mechanisms of insulin secretion. (Arq Bras Endocrinol Metab 2000;44/5: 382-389)
Keywords: Sulfonylurea receptor; SUR1 gene; Diabetes mellitus; Genetic
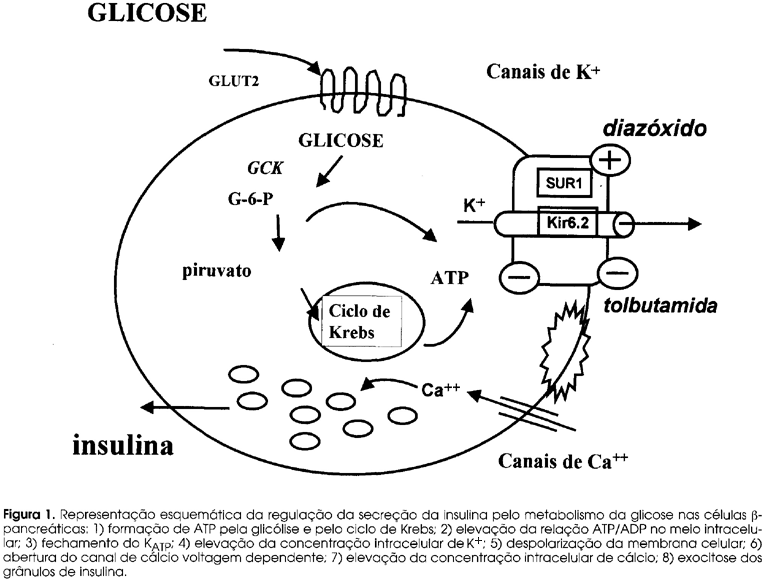
OS CANAIS IÔNICOS ESTÃO PRESENTES NA MEMBRANA plasmática e nas organelas intracelulares de todas as células, sendo responsáveis pela coordenação de diversas funções como neurotransmissão, contração, secreção e controle do volume celular. Na última década demonstrou-se que mutações nos genes codificadores para as subunidades formadoras dos canais iônicos resultam em doenças em uma enorme gama de tecidos (1). Dentre os canais iônicos, os canais de potássio ATP-dependentes expressos nas células b-pancreáticas (KATP) apresentam um papel fundamental nos mecanismos de secreção da insulina em resposta ao estímulo da glicose. (figura 1)
O KATP realiza a ligação entre o sinal gerado pelo metabolismo da glicose com a despolarização da membrana celular e com a exocitose dos grânulos de insulina (2-5). O KATP é composto de duas subunidades. A primeira, conhecida como receptor de sulfoniluréia (SUR1), é o sensor de ATP/ADP do canal. SUR1 faz parte da superfamília ABC (ATP-binding cassete) e foi assim batizada por ser o sítio de ligação desta classe de drogas utilizada no tratamento de diabéticos. A outra subunidade forma o poro do canal e é conhecida como Kir6.2 (6). Os genes codificadores para estas duas subunidades são localizados na mesma região do cromossomo 11 (p15.1) à uma distância de 4,5 kb um do outro.
Nesta revisão, procuramos discutir os principais dados da literatura que abordam o envolvimento de mutações na subunidade SUR1 do KATP em doenças tais como a hiperinsulinemia neonatal e o diabetes mellitus do tipo 2 (7).
Hipoglicemia Hiperinsulinêmica Persistente Neonatal (HHPN)
O papel importante do KATP na regulação da secreção da insulina foi evidenciado após a descoberta de que mutações nos genes codificadores para ambas as subunidades SUR1 e Kir6.2 são responsáveis pela doença conhecida como hipoglicemia hiperinsulinêmica persistente neonatal (HHPN), anteriormente conhecida como nesidioblastose (8,9). Nesta doença, observam-se níveis inapropriadamente elevados de insulina, mesmo na presença de hipoglicemia, sendo necessária a infusão de grandes quantidades de glicose para manter a euglicemia nos neonatos.
As manifestações clínicas da doença, que ocorrem predominantemente nos neonatos e crianças abaixo de um ano de idade, incluem convulsões, coma e excesso de peso para a idade gestacional. Se não for instituído o tratamento adequado podem ocorrer seqüelas neurológicas irreversíveis e mesmo a morte. A HHPN é uma doença rara com uma incidência de 1:50.000 neonatos em países ocidentais, sendo muito mais freqüente em populações com altas taxas de consangüinidade, como por exemplo na Arábia Saudita (10).
A HHPN é uma patologia heterogênea tanto do ponto de vista genético como fenotípico (11,12).
Por exemplo, duas formas histológicas da doença, focal ou difusa, podem ser observadas. A forma focal é caracterizada por uma hiperplasia nodular ou adenomatosa em uma área localizada do pâncreas. Ela representa de 30 a 40% dos casos (13). A forma difusa envolve todo o órgão, e se caracteriza por uma hiperplasia difusa das células b-pancreáticas, que se mostram irregulares, hipertrofiadas e com núcleos aumentados. A forma difusa representa a maior parte dos casos da doença (12). Clinicamente estas duas formas histológicas apresentam diferenças no que diz respeito ao início das manifestações clínicas e também à gravidade do quadro clínico. A forma difusa é mais grave, manifestando-se nas primeiras 48 horas após o nascimento, sendo necessárias ressecções extensas do pâncreas para o controle da glicemia. Na forma focal o quadro clínico é menos intenso e, em geral, a doença é identificada várias semanas após o nascimento. Nesta forma, a ressecção da lesão adenomatosa é curativa (14).
Do ponto de vista genético, as mutações do gene SUR1 constituem-se na causa mais freqüente das formas difusas de HHPN (8,10,11,15). Mais de 40 mutações foram descritas neste gene (figura 2), co-segregando com a doença com um modo de transmissão autossômico-recessivo. Muitas destas mutações descritas são associadas às formas graves da doença e demonstram in vitro uma ausência da atividade do KATP. Ressalta-se que um efeito genético fundador parece estar presente em certas populações (presença de um ancestral comum). Por exemplo, duas mutações (3992-9G->A e DF 1388) representam 88% dos casos de HHPN nos judeus Askenazes (10), e a mutação V187D representa a maior parte dos casos da doença na Finlândia (15). Da mesma forma, observou-se que mutações no gene codificador para a outra subunidade do KATP (Kir6.2) são responsáveis por alguns casos da forma difusa de HHPN, mas com freqüência bem menor se comparadas às mutações no gene SUR1 (8,15).
Do ponto de vista fisiopatológico, as mutações em ambos os genes (SUR1 e Kir6.2) promovem uma perda da atividade do KATP levando a um estado de estimulação permanente da secreção da insulina, sem regulação pelos níveis circulantes de glicose. Outros casos familiares de HHPN, cuja transmissão se faz de uma forma autossômica dominante, são associados às mutações raras de genes, como o da glicoquinase, glutamato-desidrogenase e também de outros loci ainda não identificados (18-20).
Além das formas familiares de HHPN, existem algumas formas esporádicas focais cujos mecanismos moleculares foram elucidados recentemente (13,21). O caso de gêmeos monozigotos discordantes para a doença sugeriu que as mutações constitucionais homozigóticas do gene SUR1 não eram as únicas responsáveis pelo estado de hiperinsulinemia (13). Demonstrou-se que o defeito molecular resultava da associação de uma mutação heterozigótica do gene SUR1, de origem paterna, e de um fenômeno epigenético a nível pancreático. Este fenômeno se caracteriza por uma perda de alelos maternos da região 11p15, conduzindo a hemizigose ou a homozigose da mutação do gene SUR1 de origem paterna no tecido tumoral. A origem da formação tumoral parece estar também associada a esta perda de alelos maternos. De fato, a região excluída contém genes supressores do crescimento tecidual, cuja expressão é restrita aos alelos maternos (fenômeno conhecido sob o nome de "imprinting") (21,22). Desta forma, a ausência dos alelos maternos conduz, de maneira independente, à formação do tumor e ao defeito de secreção de insulina.
KATP e Diabetes Mellitus do Tipo 2
O envolvimento dos genes SUR1 e Kir6.2 nos mecanismos fisiopatológicos da HHPN, demonstrou o papel regulador destes genes na secreção de insulina. Esta observação conduziu vários pesquisadores a estudar um possível papel destes genes na susceptibilidade genética ao diabetes mellitus tipo 2 (DM2). Inúmeras variantes foram identificadas no gene SUR1, entre as quais um polimorfismo intrônico próximo ao exon 16 (-3c->t; cagGCC->tagGCC), assim como uma mutação silenciosa localizada no exon 18 (ACC->ACT; Thr759Thr) (23). Estas duas variantes foram estudadas em várias populações de diabéticos. Encontrou-se associação entre ambas as variantes e DM2 em ingleses e americanos de origem mexicana (23). Em diabéticos holandeses e finlandeses, encontrou-se uma associação da doença com a variante no exon 16, mas não com a do exon 18 (24,25). Interessante salientar que nos estudos de populações de diabéticos ingleses e americanos a associação foi observada com o alelo cagGCC da variante no exon 16, enquanto que nos estudos nos holandeses e finlandeses a associação encontrada foi com o alelo tagGCC da mesma variante. Por outro lado, em pacientes franceses selecionados por apresentar forte história familiar de DM2, encontrou-se uma associação com a variante do exon 18 e não com a do exon 16 deste gene (26). Neste estudo, a associação da variante no exon 18 com o DM2 pareceu ser dependente do sobrepeso presente nos indivíduos diabéticos. Neste mesmo estudo, a análise em uma população de franceses obesos mórbidos confirmou a associação da variante no exon 18 com a obesidade (26). Os autores também observaram que os obesos homozigotos para o alelo tagGCC da variante no exon 16 apresentavam uma obesidade mais grave em comparação aos portadores dos outros genótipos.
Nós estudamos estas duas variantes em outro grupo de diabéticos tipo 2 franceses, não selecionados pela história familiar de DM, e não encontramos associações com a doença ou com o índice de massa corporal. Em relação à variante no exon 18, observamos uma freqüência do alelo T de 5,7% nos diabéticos em comparação à 2,8% nos controles, uma diferença não significativa em função do tamanho da amostra (27). Os resultados discordantes acima descritos revelam o caráter multigênico do DM2 onde alelos de risco diferentes são encontrados em populações e grupos de diabéticos diversos.
Variantes do Gene SUR1 e Secreção de Insulina
Na literatura, poucos estudos abordaram o padrão de secreção da insulina nos portadores das diferentes variantes alélicas do gene SUR l. Em um estudo em dinamarqueses não diabéticos, a secreção da insulina em resposta ao estímulo com glicose intravenosa ou tolbutamida foi semelhante nos portadores dos diferentes alelos das variantes nos exons 16 e 18 (28). Entretanto, neste estudo, os indivíduos que eram ao mesmo tempo homozigotos para o alelo tagGCC do exon 16 e heterozigotos para o alelo T do exon 18, apresentavam uma redução da secreção de insulina e do peptídeo C após estímulo com a tolbutamida, (figura 3)
Em um estudo recente realizado em holandeses não diabéticos ou intolerantes à glicose, demonstrou-se que os indivíduos portadores do alelo tagGCC do exon 16 apresentavam redução da segunda fase de secreção da insulina (29). Neste estudo, a variante no exon 18 não foi associada a diferenças no padrão de secreção da insulina.
Nós estudamos recentemente o padrão de secreção da insulina, da depuração metabólica da glicose e da sensibilidade à insulina em indivíduos não diabéticos portadores dos diferentes alelos do exon 18, todos parentes de primeiro grau de diabéticos tipo 2. Os parâmetros analisados foram semelhantes entre os grupos (Reis AF e cols, dados não publicados), sugerindo que a variante do exon 18 deve apresentar um impacto bastante limitado no padrão de secreção da insulina. No entanto, os dados não descartam a hipótese da associação desta variante com a doença, ou de uma outra variante localizada na sua proximidade que seja transmitida em conjunto. De fato, um estudo familiar realizado em americanos de origem mexicana, demonstrou uma associação entre a glicemia de 2 horas após sobrecarga oral com a glicose, com marcadores genéticos localizados na proximidade do locus do gene SUR1 (30). Mais recentemente descreveu-se a associação entre uma mutação silenciosa no exon 31 do gene SUR1 (AGG®AGA, Arg1272Arg) com os níveis de insulina de jejum e após sobrecarga oral com a glicose em indivíduos não diabéticos americanos de origem mexicana (31). Neste estudo, os indivíduos portadores do genótipo AA apresentavam níveis de insulina significativamente superiores em comparação àqueles portadores dos outros genótipos (AG ou GG). Estes dados nos estimularam a procurar estabelecer uma possível associação desta variante com o DM2 em franceses caucasianos (27). Encontramos uma associação do alelo A com a doença (odds ratio:1,37 à 2,92). Esta associação foi mais evidente naqueles diabéticos cujo diagnóstico havia sido feito antes dos 45 anos de idade. Em um estudo finlandês, descreveu-se uma associação do outro alelo desta variante (alelo G) com diabetes gestacional (25). No entanto, neste estudo, o padrão de secreção de insulina em resposta ao estímulo oral ou intravenoso com glicose foi semelhante nos portadores não diabéticos dos diferentes alelos.
Lições do Camundongo "Knockout" para o Gene SUR1
Os modelos animais com construção de "knockout" em determinado gene são de grande utilidade para a compreensão das repercussões fenotípicas da perda da função deste gene. Recentemente, Seghers e cols (32) construíram camundongos homozigóticos com perda do gene SUR1(SUR1 -/-).Estes animais são viáveis e férteis, mas são anormais quanto à secreção de insulina. Os camundongos neonatos SUR1 -/-são hipoglicêmicos um dia após o nascimento, com uma relação insulina/glicose inapropriadamente alta. Esta hipoglicemia é revertida no segundo dia de vida, e no quinto dia os animais SUR1 -/-mostram-se hiperglicêmicos, com uma relação insulina/glicose que é diminuída pela metade em comparação aos animais SUR1 +/+.
Os animais SUR1 -/-apresentam intolerância a glicose com níveis reduzidos de secreção da insulina, após uma sobrecarga de glicose intraperitoneal. No entanto, após jejum prolongado de 16 horas os animais SUR1 -/-são mais hipoglicêmicos do que os controles, o que é consistente com a observação feita da incapacidade das ilhotas SUR1 -/- de diminuir a secreção de insulina quando se reduzem os níveis de glicose. Após a infusão contínua de glicose por 8 horas, os animais SUR1 -/-apresentam taxas de secreção de insulina inferiores em comparação aos controles. No entanto, após o término do estímulo, a secreção de insulina dos animais SUR1 -/-tarda mais a retornar aos níveis basais, resultando em uma hiperinsulinemia residual. Os animais não apresentam modificação da sensibilidade à insulina.
Do ponto de vista histológico, as ilhotas dos animais adultos são normais e não apresentam uma taxa maior de apoptose em relação ao grupo controle. Nas ilhotas isoladas dos animais SUR1 -/- observa-se uma perda da primeira fase de secreção da insulina após estímulo com glicose e uma segunda fase atenuada em comparação às ilhotas controle.
Como comentado acima, indivíduos portadores do alelo tagGCC do exon 16 apresentam redução da segunda fase de secreção da insulina (29). As ilhotas dos animais SUR1 -/- também apresentavam uma ausência de resposta ao estímulo com a tolbutamida, o que está de acordo com o estudo realizado em humanos nos portadores das variantes nos exons 16 e 18 (28). Este estudo corrobora o papel da subunidade SUR1 nos mecanismos de secreção insulínica, e de seu possível envolvimento nos mecanismos de hiperglicemia.
CONCLUSÕES
A análise dos dados atualmente disponíveis na literatura sugere que SUR1 não apresentaria um papel de "gene maior" na susceptibilidade genética ao DM2 (30,34). No entanto, parece certo que o gene SUR1, ou um outro gene nesta mesma região do cromossomo 11p15.1, apresenta um papel de "gene menor ou modificador" dentro do contexto poligênico da doença (7). Ressalta-se que as variantes de SUR1 que foram até então estudadas, são polimorfismos intrônicos ou mutações silenciosas que, em princípio, não modificam a proteína. Especula-se que haja um desequilíbrio de transmissão destas variantes com alguma mutação funcional nas regiões codificadoras do gene ou mesmo com sua região promotora. Esta região, segundo nosso conhecimento, ainda não foi estudada.
Por outro lado, as diferentes distribuições alélicas, descritas em diversas populações, confirmam o caráter multigênico do DM2, onde alelos de risco distintos são encontrados em grupos diversos. Esta característica poderia ser explicada por graus variáveis de desequilíbrio de transmissão das variantes com mutações funcionais em diferentes populações. Uma outra hipótese seria o envolvimento de um outro gene da região 11p15.1 na associação com a doença. O gene codificador para a outra subunidade do KATP , o Kir6.2 é um candidato natural. O "knockout" deste gene em camundongos promove um estado de hiperglicemia, com alteração do padrão de secreção da insulina assim como um aumento da apoptose nas células b-pancreáticas (35,36). Na espécie humana descreveu-se inúmeras variantes deste gene (37,38). Em um estudo realizado em dinamarqueses não diabéticos, demonstrou-se diferentes padrões de sensibilidade à insulina relacionados à diversos haplotipos deste gene, com níveis de secreção de insulina semelhantes (39). Hani e cols (39), realizaram uma meta-análise de resultados em várias populações e sugeriram a existência de associação de uma variante do gene Kir6.2 (Glu23Lys) com o DM2. Ressalta-se que esta variante parece não modificar a função do canal em ensaios in vitro (40).
Em um estudo francês, demonstrou-se que esta variante do gene Kir6.2 não estava em desequilíbrio de transmissão com as variantes dos exons 16 ou 18 do gene SUR1 (39). Este resultado não descarta um possível desequilíbrio de transmissão de variantes do gene Kir6.2 com regiões promotoras do próprio gene ou do gene SUR1, regiões ainda não exploradas.
Em resumo, o papel do KATP nos mecanismos de secreção da insulina é bem estabelecido. Mutações nos genes codificadores para as duas subunidades do canal (SUR1 e Kir6.2) são responsáveis por grande parte dos casos de HHPN. Em relação ao DM2, estes dois genes parecem não ser genes maiores para a susceptibilidade à doença, sendo, provavelmente, moduladores de fenótipos associados à doença (secreção da insulina, sensibilidade à insulina e obesidade). Desta forma, variantes destes genes podem contribuir ao determinismo genético do DM2 dentro de um contexto multifatorial e poligenético. A identificação destas variantes funcionais permitirá um diagnóstico fármaco-genético dos diabéticos tipo 2 e a instituição de um tratamento precoce e mais específico para estes indivíduos.
AGRADECIMENTOS
Durante a realização dos trabalhos citados nas referências 7 e 27, A.F. Reis foi beneficiado com uma bolsa de doutorado concedida pelo CAPES (processo 0473/98-7), Brasília, Brasil.
7. Reis AF, Velho G. Pathologie moléculaire du récepteur des sulfamides SUR1 et altérations de l'insulinosécrétion. Méd Thérap Endocrinol (no prelo).
27. Reis AF, Ye WZ, Dubois D, Bellanné-Chantelot C, Timsit J, Velho G. Association of a variant in exon 31 of the sulfonylurea receptor 1 (SUR1) gene with type 2 diabetes mellitus in French Caucasians. Hum Genet 2000 (no prelo).
Endereço para correspondência:
Gilberto Velho
INSERM U-342, Hôpital Saint-Vincent-de-Paul,
82, Avenue Denfert Rochereau, 75014 Paris, França
Fone: 33-01 40 48 82 48, FAX: 33 1 40 48 83 52
e-mail: gvelho@infobiogen.fr
- 1. Aguilar-Bryan L, Bryan J. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr Rev 1999;20:101-35.
- 2. Cook DL, Hales CN. Intracellular ATP directly blocks K+ channel in pancreatic beta cells. Nature 1984;311:271-3.
- 3. Dukes ID, Philipson LH. K+ channels: generating excitement in pancreatic b-cells. Diabetes 1996;45:845-53.
- 4. Inagaki N, Seino S. ATP-sensitive potassium channels: Structures, functions, and pathophysiology. Jpn J Physiol 1998;48:397-412.
- 5. Ashcroft FM, Gribble FM. ATP-sensitive K+ channels and insulin secretion: their role in health and disease. Diabetologia 1999;42:903-19.
- 6. Inagaki N, Gonoi T, Clement IV JP, Namba N, Inazawa J, Gonzales G, et al. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science 1995;270:1166-70.
- 8. Thomas PM, Cote GJ, Wohlik N, Haddad B, Mathew PM, Rabl W, et al. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science 1995;268:426-9.
- 9. Thomas P, Ye YY, Lightner E. Mutation of the pancreatic islet inward rectifier kir6.2 also leads to familial persistent hyperinsulinemic hypoglycemia of infancy. Hum Mol Genet 1996;5:1809-12.
- 10. Nestorowicz A, Wilson BA, Kathleen PS, Inoue H, Glaser B, Landau H, et al. Mutations in the sulfonylurea receptor gene are associated with familial hyperinsulinism in Ashkenazi jews. Hum Mol Genet 1996;5:1813-22.
- 11. Nestorowicz A, Glaser B, Wilson BA, Shyng SL, Nichols CG, Stanley CA, et al. Genetic heterogeneity in familial hyperinsulinism. Hum Mol Genet 1998;7:1119-28.
- 12. de Lonlay-Debeney P, Poggi-Travert F, Fournet JC, Sempoux C, Vici CD, Brunelle F, et al. Clinical features of 52 neonates with hyperinsulinism. N Engl J Med 1999;340:1169-75.
- 13. Bonnefont J, de Lonlay P, Fournet J, Gross-Morand M, Poggi-Travert F, Foussier V, et al. Somatic deletion of imprinted 11p15 region in sporadic persistent hyperinsulinemic hypoglycemia of infancy is specific of focal adenomatous, hyperplasía and endorses partial pancreatectomy. J Clin Invest 1997;100:802-7.
- 14. Sempoux C, Poggi F, Brunelle F, Saudubray JM, Fekete C, Rahier J. Nesidioblastosis and persistent neonatal hyperinsulinism. Diab Metab 1995;21:402-7.
- 15. Otonkoski T, Ammala C, Huopio H, Cote GJ, Chapman J, Cosgrove K, et al. A point mutation inactivating the sulfonylurea receptor causes the severe form of persistent hyperinsulinemic hypoglycemia of infancy in Finland, Diabetes 1999;48:408-15.
- 16. Nestorowicz A, Inagaki N, Tohru G, Schoor KP, Wilson BA, Glaser B, et al. 1997 A nonsense mutation in the inward rectifier potassium channel gene, Kir6.2, is associated with familial hyperinsulinism. Diabetes 1997;46:1743-8.
- 17. Glaser B, Kesavan P, Heyman M, Davis E, Cuesta A, Buchs A, et al. Familial hyperinsulinism caused by an activating glucokinase mutation. N Engl J Med 1998;338:226-30.
- 18. Stanley C, Lieu YK, Hsu BY, Burlina AB, Greenberg CR, Hopwood NJ, et al. Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N Engl J Med 1998;338:1352-7.
- 19. Kukuvitis A, Deal C, Arbour L, Polychronakos C. An autosomal dominant form of familial persistent hyperinsulinemic hypoglycemia of infancy, not linked to the sulfonylurea receptor locus. J Clin Endocrinol Metab 1997;82:1192-4,
- 20. Verkarre V, Fournet JC, de Lonlay P, Gross-Morand MS, Devillers M, Rahier J, et al. Paternal mutation of the sulfonylurea receptor (SUR1) gene and maternal loss of 11p15 imprinted genes lead to persistent hyperinsulinism in focal adenomatous hiperplasia. J Clin Invest 1998;102:1286-91.
- 21. Hark AT, Schoenherr J, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation-sensitive enhancer-blockíng activity at the H19/lgf2 locus. Nature 2000;405:486-9.
- 22. Bell AC, Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2gene. Nature 2000;405:482-5.
- 23. Inoue H, Ferrer J, Welling CM, Elbein SC, Hoffman M, Mayorga R, et al. Sequence variants in the sulfonylurea receptor (SUR) gene are associated with NIDDM in Caucasians. Diabetes 1996;45:825-31.
- 24. 't Hart LM, de Knijff P, Dekker JM, Stolk RP, Nijpels G, van der Does FEE, et al. Variants in the sulfonylurea receptor gene: association of the exon 16-3t variant with Type II diabetes mellitus in Dutch Caucasians. Diabetologia 1999;42:617-20.
- 25. Rissanen J, Markkanen A, Karkkainen P, Pihlajamaki J, Kekalainen P, Mykkanen L, et al. Sulfonylurea receptor-1 gene variants are associated with gestational diabetes and type 2 diabetes but not with altered secretion of insulin. Diabetes Care 2000;23:70-3.
- 26. Hani EH, Clément K, Velho G, Vionnet N, Hager J, Philippi A, et al. Genetic studies of the sulfonylurea receptor gene locus in NIDDM and in morbid obesity among French Caucasians. Diabetes 1997;46:688-94.
- 28. Hansen T, Soren ME, Hansen L, Moller AM, Almind K, Clausen JO, et al. Decreased tolbutamide-stimulated insulin secretion in healthy subjects with sequence variants in the high-affinity sulfonylurea receptor gene. Diabetes 1998;47:598-605.
- 29. 't Hart LM, Dekker JM, van Haeften TW, Ruige JB, Stehouwer CDA, Erkelens DW, et al. Reduced second phase insulin secretion in carriers of a sulphonylurea receptor gene variant associating with type II diabetes mellitus. Diabetologia 2000;43:515-9.
- 30. Lindner T, Gragnoli C, Schulze J, Rietzsch H, Petzold C, Schroder H-E, et al. The 31-cM region of chromosome 11 including the obesity gene tubby and ATP-sensitive potassium channel genes, SUR1 and Kir6.2, does not contain a major susceptibility locus for NIDDM in 127 non-Hispanic white affected sibships. Diabetes 1997;46:1227-9.
- 31. Goksel DL, Rschbach K, Duggirala R, Mitchell BD, Aguilar-Bryan L, Blangero J, et al. Variant in sulfonylurea receptor-1 gene is associated with high insulin concentrations in non-diabetic Mexican Americans: SUR-1 gene variant and hyperinsulinemia. Hum Genet 1998;103:280-5.
- 32. Seghers V, Nakazaki M, DeMayo F, Aguilar-Bryan L, Bryan J. SUR1 knockout mice. A model for KATP channel-independent regulation of insulin secretion. J Biol Chem 2000;275:9279-87.
- 33. Iwasaki N, Kawamura M, Yamagata K, Cox NJ, Karibe S, Ohgawara H, et al. Identification of microsatellite markers near the human genes encoding the beta-cell ATP-sensitive k+ channel and linkage studies with NIDDM in Japanese. Diabetes 1996;45:267-9.
- 34. Stirling B, Cox NJ, Bell Gl, Hanis CL, Spielman RS, Concannon P. Linkage studies in NIDDM with markers near the sulphonylurea receptor gene. Diabetologia 1995;38:1479-81.
- 35. Miki T, Tashiro F, Iwanaga T, Nagashima K, Yoshitomi H, Aihara H, et al. Abnormalities of pancreatic islets by targeted expression of a dominant-negative KATP channel. Proc Natl Acad Sci USA 1997;94:11969-73.
- 36. Miki T, Nagashima K, Tashiro F, Kotake K, Yoshitomi H, Tamamoto A, et al. Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc Natl Acad Sci USA 1998;95:10402-6.
- 37. Inoue H, Ferrer J, Warren-Perry M, Zhang Y, Millns H, Turner RC, et al. Sequence variants in the pancreatic islet beta-cell inwardly rectifying K+ channel Kir6.2 (Bir) gene: Identification and lack of role in Caucasian patients with NIDDM. Diabetes 1997;46:502-7.
- 38. Hansen L, Echwald SM, Hansen T, Urhammer SA, Clausen JC, Pedersen O. Amino acid polymorphisms in the ATP-regulatable inward rectifier Kir6.2 and their relationships to glucose- and tolbutamide-induced insulin secretion, the insulin sensitivity index, and NIDDM. Diabetes 1997;46:508-12.
- 39. Hani EH, Boutin P, Durand E, Inoue H, Permutt MA, Velho G, et al. Missense mutations in the pancreatic islet beta cell inwardly rectifying K+ channel gene (KIR6.2/BIR): a meta-analysis suggest a role in the polygenic basis of type II diabetes mellitus in Caucasians. Diabetologia 1998;41:1511-5.
- 40. Sakura H, Wat N, Horton V, Millns H, Turner RC, Ashcroft FM. Sequence variations in the human Kir6.2 gene, a subunit of the beta-cell ATP-sensitive k-channel: no association with NIDDM in white caucasian subjects or evidence of abnormal function when expressed in vitro. Diabetologia 1996;39:1233-6.
Datas de Publicação
-
Publicação nesta coleção
28 Set 2005 -
Data do Fascículo
Out 2000
Histórico
-
Aceito
15 Ago 2000 -
Recebido
08 Ago 2000