Resumos
INTRODUÇÃO: Sintomas musculoesqueléticos podem representar a primeira manifestação de imunodeficiências humorais primárias. A freqüência de deficiência seletiva de IgA em pacientes com artrite idiopática juvenil (AIJ símile) e lúpus eritematoso sistêmico juvenil (LESJ) é de 2% a 4% e de 1% a 4%, respectivamente. OBJETIVO: Descrever pacientes que apresentaram artrite como primeiro sinal de uma imunodeficiência humoral primária e determinar a prevalência de deficiência seletiva de IgA em pacientes com diagnóstico de AIJ e LESJ. PACIENTES E MÉTODOS: Entre janeiro de 1983 e dezembro de 2006, 4.876 pacientes foram acompanhados na Unidade de Reumatologia Pediátrica. Uma avaliação retrospectiva foi realizada em pacientes que apresentaram artrite como primeira manifestação de imunodeficiência. As imunodeficiências humorais foram classificadas em: deficiência seletiva de IgA, hipogamaglobulinemia e deficiência de subclasses de IgG. RESULTADOS: Onze (0,2%) pacientes apresentaram imunodeficiências humorais: deficiência seletiva de IgA ocorreu em oito, imunodeficiência comum variável em dois e deficiência de subclasses de IgG em um. Cinco dos 11 pacientes apresentaram artrite aguda e seis apresentaram artrite crônica não-erosiva (AIJ símile). Dosagem de imunoglobulinas foi realizada em 70 dos 253 pacientes com AIJ e deficiência seletiva de IgA (IgA sérica < 7 mg/dL) foi detectada em 6 (8,5%) - AIJ símile. Dos 45 pacientes com LESJ, com dosagem de IgA realizada, 3 (6,6%) apresentaram deficiência seletiva de IgA. CONCLUSÃO: O presente estudo descreveu baixa prevalência de imunodeficiências humorais primárias em pacientes com doenças reumatológicas. Entretanto, a associação entre doenças auto-imunes e imunodeficiências sugere semelhanças em sua etiopatogenia e deve incentivar estudos prospectivos para investigação desta hipótese.
artrite; artrite idiopática juvenil; lúpus eritematoso sistêmico juvenil; imunodeficiência
INTRODUCTION: Rheumatologic findings may be the first manifestation of primary humoral immunodeficiencies. The frequency of selective IgA deficiency in patients with juvenile idiopathic arthritis (JIA like) and juvenile systemic lupus erithematosus (JSLE) is 2% to 4% and 1% to 4%, respectively. OBJECTIVE: To describe patients with primary humoral immunodeficiencies associated with arthritis and to determine the prevalence of selective IgA deficiency within JIA and JSLE patients. PATIENTS AND METHODS: From January 1983 to December 2006, 4.876 patients were followed at the Pediatric Rheumatology Unit. A retrospective evaluation was performed in patients that presented arthritis as the first clinical manifestation of immunodeficiency. The humoral immunodeficiencies were classified into selective IgA deficiency, hypogammaglobulinemia and IgG subclass deficiency. RESULTS: Eleven patients (0.2%) had primary immunodeficiency: selective IgA deficiency occurred in 8, common variable immunodeficiency in two, and IgG subclass deficiency in one. Five of the 11 patients had an acute arthritis and six patients a chronic nonerosive arthritis (JIA-like). From the 253 JIA patients evaluated, 70 had IgA level evaluation done and 6 (8.5%) presented complete IgA deficiency (serum IgA < 7 mg/dl) (JIA-like). From the 45 JSLE patients with IgA levels evaluated, 3 (6.6%) had selective IgA deficiency diagnosis. CONCLUSION: The present study showed a low prevalence of humoral immunodeficiency in patients with rheumatic diseases. However, this association suggests that similar defects in immune response could be related to both diseases and that prospective studies are needed to elucidate this hypothesis.
arthritis; juvenile idiopathic arthritis; juvenile systemic lupus erythematosus; immunodeficiency
ARTIGO ORIGINAL ORIGINAL ARTICLE
Associação de imunodeficiências primárias com doenças auto-imunes na infância
Primary immunodeficiencies and autoimmune diseases association in childhood
Adriana Almeida de JesusI; João Carlos DinizII; Bernadete de Lourdes LiphausIII; Cristina Miuki Abe JacobIV; Magda Carneiro-SampaioV; Clovis Artur Almeida da SilvaVI
IAluna da pós-graduação do Departamento de Pediatria da FMUSP
IIMédico da Complementação Especializada da Unidade de Reumatologia Pediátrica do ICr-HC-FMUSP
IIIDoutora em Ciências pela FMUSP e médica-assistente da Unidade de Reumatologia Pediátrica do Departamento de Pediatria da FMUSP
IVDoutora em Medicina pela FMUSP e responsável pela Unidade de Alergia e Imunologia Pediátrica do Departamento de Pediatria da FMUSP
VProfessora titular do Departamento de Pediatria da FMUSP
VIProfessor livre-docente do Departamento de Pediatria da FMUSP e responsável pela Unidade de Reumatologia Pediátrica do Departamento de Pediatria da FMUSP
Endereço para correspondência Endereço para correspondência: Adriana Almeida de Jesus Rua Oscar Freire, 2040/124, Jardim América, CEP 05409-011, São Paulo, SP e-mail: adriaj@uol.com.br
RESUMO
INTRODUÇÃO: Sintomas musculoesqueléticos podem representar a primeira manifestação de imunodeficiências humorais primárias. A freqüência de deficiência seletiva de IgA em pacientes com artrite idiopática juvenil (AIJ símile) e lúpus eritematoso sistêmico juvenil (LESJ) é de 2% a 4% e de 1% a 4%, respectivamente.
OBJETIVO: Descrever pacientes que apresentaram artrite como primeiro sinal de uma imunodeficiência humoral primária e determinar a prevalência de deficiência seletiva de IgA em pacientes com diagnóstico de AIJ e LESJ.
PACIENTES E MÉTODOS: Entre janeiro de 1983 e dezembro de 2006, 4.876 pacientes foram acompanhados na Unidade de Reumatologia Pediátrica. Uma avaliação retrospectiva foi realizada em pacientes que apresentaram artrite como primeira manifestação de imunodeficiência. As imunodeficiências humorais foram classificadas em: deficiência seletiva de IgA, hipogamaglobulinemia e deficiência de subclasses de IgG.
RESULTADOS: Onze (0,2%) pacientes apresentaram imunodeficiências humorais: deficiência seletiva de IgA ocorreu em oito, imunodeficiência comum variável em dois e deficiência de subclasses de IgG em um. Cinco dos 11 pacientes apresentaram artrite aguda e seis apresentaram artrite crônica não-erosiva (AIJ símile). Dosagem de imunoglobulinas foi realizada em 70 dos 253 pacientes com AIJ e deficiência seletiva de IgA (IgA sérica < 7 mg/dL) foi detectada em 6 (8,5%) AIJ símile. Dos 45 pacientes com LESJ, com dosagem de IgA realizada, 3 (6,6%) apresentaram deficiência seletiva de IgA.
CONCLUSÃO: O presente estudo descreveu baixa prevalência de imunodeficiências humorais primárias em pacientes com doenças reumatológicas. Entretanto, a associação entre doenças auto-imunes e imunodeficiências sugere semelhanças em sua etiopatogenia e deve incentivar estudos prospectivos para investigação desta hipótese.
Palavras-chave: artrite, artrite idiopática juvenil, lúpus eritematoso sistêmico juvenil, imunodeficiência.
ABSTRACT
INTRODUCTION: Rheumatologic findings may be the first manifestation of primary humoral immunodeficiencies. The frequency of selective IgA deficiency in patients with juvenile idiopathic arthritis (JIA like) and juvenile systemic lupus erithematosus (JSLE) is 2% to 4% and 1% to 4%, respectively.
OBJECTIVE: To describe patients with primary humoral immunodeficiencies associated with arthritis and to determine the prevalence of selective IgA deficiency within JIA and JSLE patients.
PATIENTS AND METHODS: From January 1983 to December 2006, 4.876 patients were followed at the Pediatric Rheumatology Unit. A retrospective evaluation was performed in patients that presented arthritis as the first clinical manifestation of immunodeficiency. The humoral immunodeficiencies were classified into selective IgA deficiency, hypogammaglobulinemia and IgG subclass deficiency.
RESULTS: Eleven patients (0.2%) had primary immunodeficiency: selective IgA deficiency occurred in 8, common variable immunodeficiency in two, and IgG subclass deficiency in one. Five of the 11 patients had an acute arthritis and six patients a chronic nonerosive arthritis (JIA-like). From the 253 JIA patients evaluated, 70 had IgA level evaluation done and 6 (8.5%) presented complete IgA deficiency (serum IgA < 7 mg/dl) (JIA-like). From the 45 JSLE patients with IgA levels evaluated, 3 (6.6%) had selective IgA deficiency diagnosis.
CONCLUSION: The present study showed a low prevalence of humoral immunodeficiency in patients with rheumatic diseases. However, this association suggests that similar defects in immune response could be related to both diseases and that prospective studies are needed to elucidate this hypothesis.
Keywords: arthritis, juvenile idiopathic arthritis, juvenile systemic lupus erythematosus, immunodeficiency.
INTRODUÇÃO
As doenças reumatológicas da faixa etária pediátrica podem estar relacionadas com uma grande variedade de causas e diversas manifestações musculoesqueléticas e podem representar a primeira manifestação clínica de uma imunodeficiência primária(1).
A presença de artrite nas imunodeficiências humorais primárias é raramente descrita na literatura e pode ser a primeira e única manifestação clínica até que o diagnóstico seja estabelecido(2). A freqüência de deficiência seletiva de IgA (Def IgA) em pacientes com artrite crônica [artrite idiopática juvenil (AIJ) símile] é descrita entre 2% e 4%(3-5) dos casos, e o percentual da prevalência dessa imunodeficiência em pacientes com lúpus eritematoso sistêmico juvenil (LESJ) é de 1% a 4%(3,6,7). A associação de doenças reumatológicas com hipogamaglobulinemia também é descrita com freqüências que variam de 7% a 42%(3). Por outro lado, pacientes com imunodeficiência comum variável (IDCV) ou Def IgA apresentam maior prevalência de AIJ, LESJ e síndrome de Sjögren(8).
A raridade de estudos de prevalência dessa associação, particularmente na população brasileira de AIJ e LESJ, e a expressiva casuística da Unidade de Reumatologia Pediátrica do Instituto da Criança do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (ICr- HC-FMUSP) estimularam a realização desta pesquisa.
Portanto, os objetivos do presente estudo foram descrever os pacientes que apresentaram artrite como manifestação inicial de imunodeficiências humorais primárias e avaliar a prevalência de Def IgA na população de pacientes com AIJ e LESJ, acompanhados nesse serviço terciário de reumatologia pediátrica.
PACIENTES E MÉTODOS
O estudo realizado foi transversal. Durante o período de 23 anos (janeiro de 1983 a dezembro de 2006), 4.876 pacientes foram acompanhados na Unidade de Reumatologia do ICr-HC-FMUSP e foram avaliados aqueles que apresentaram artrite como primeira manifestação de imunodeficiências humorais primárias. A avaliação retrospectiva dos prontuários médicos incluiu o registro de dados demográficos (sexo, idade de início da doença reumatológica e idade de diagnóstico da imunodeficiência primária), história de doença auto-imune ou imunodeficiências em parentes de primeiro e segundo graus, história de infecções de repetição, descrição detalhada da doença articular (padrão de início, duração dos sintomas, número de articulações comprometidas, distribuição do envolvimento articular, localização e seqüência de acometimento das articulações afetadas) e presença de manifestações extra-articulares.
As imunodeficiências humorais avaliadas no presente estudo foram: Def IgA (idade acima de 4 anos, níveis séricos de IgA < 7 mg/dL e níveis normais de IgG e IgM); hipogamaglobulinemia, que pode ser classificada em três subtipos: agamaglobulinemia ligada ao X [redução de todos os isotipos de imunoglobulinas (IgG < 2 g/L, IgA e IgM < 0,2 g/L) e linfócitos B ausentes ou muito reduzidos (< 2%)], IDCV (redução dos níveis de IgG abaixo de dois desvios-padrão para a faixa etária, níveis séricos IgA reduzidos, IgM usualmente normal e linfócitos B em número normal ou discretamente reduzidos) e hipogamaglobulinemia transitória da infância (redução dos níveis de IgG e IgA com número normal de linfócitos B, que ocorre antes dos 3 anos de idade) e deficiência de subclasses de IgG [redução em uma ou mais subclasses de IgG (1, 2, 3 e/ou 4), abaixo de dois desvios-padrão da média para a faixa etária, com número normal de linfócitos B)(9).
A avaliação laboratorial de auto-imunidade incluiu dosagem de fator reumatóide e de auto-anticorpos: fator antinúcleo (FAN), anti-DNA nativo, anti-Sm, anti-Ro/SSA, anti-La/SSB, anticardiolipina (isotipos IgG e IgM) e anticoagulante lúpico. Outros auto-anticorpos, avaliados nos pacientes com LESJ para pesquisa de auto-imunidade órgão-específica, foram: tireóide (antiperoxidase, antitireoglobulina e anti-receptor de TSH TRAB), fígado (antimitocondrial, antimicrossomal, antimúsculo liso e anticitosol hepático), trato gastrintestinal (anticélula parietal, antigliadina IgG e anti-endomísio) e diabetes melito (anti-insulina e antidescarboxilase do ácido glutâmico anti-GAD). Radiografias das articulações acometidas foram também realizadas para pesquisa de osteopenia periarticular, erosões, cistos ósseos e anquilose.
O diagnóstico de LESJ foi estabelecido de acordo com os critérios de classificação do American College of Rheumatology (ACR), com início da doença antes dos 18 anos de idade(10). Os critérios do International League of Associations for Rheumatology (ILAR) foram utilizados para o diagnóstico de AIJ(11). O presente estudo foi aprovado pelo Comitê de Pesquisa e Ética do HC-FMUSP.
RESULTADOS
Ao longo de 23 anos, 4.876 pacientes foram acompanhados na Unidade de Reumatologia do ICr-HC-FMUSP. Destes, 620 pacientes tiveram diagnóstico de AIJ e 244, de LESJ.
Dos 620 pacientes que tiveram diagnóstico de AIJ, os prontuários de 253 foram avaliados e 70 (11%) tinham dosagens séricas de imunoglobulinas (IgG, IgA e IgM). A deficiência de IgA foi encontrada em 6/70 (8,5%) dos pacientes com AIJ (AIJ símile). Dos 244 pacientes com LESJ avaliados neste período, 45 (18%) realizaram dosagens séricas de imunoglobulinas, e deficiência de IgA foi encontrada em 3/45 (6,6%) dos pacientes com LESJ.
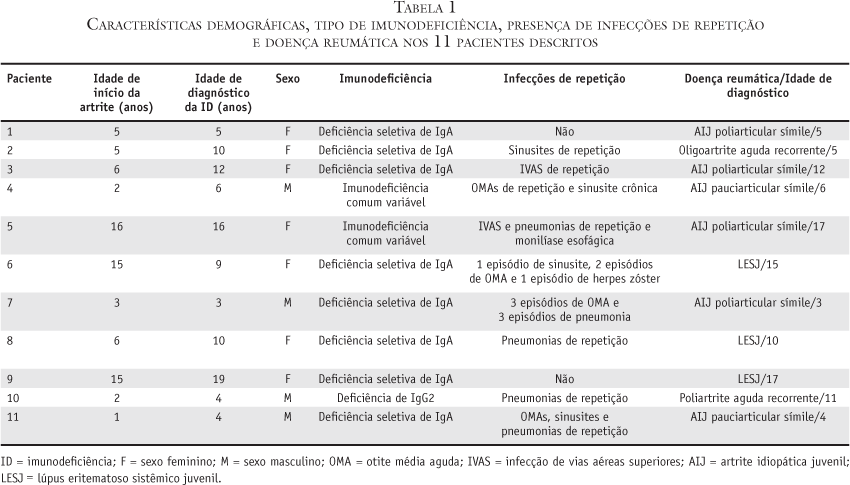
O diagnóstico de imunodeficiência humoral foi realizado retrospectivamente em 11 pacientes (0,2%), incluindo os seis pacientes com AIJ símile e os três com LESJ. A tabela 1 inclui características demográficas, tipo de imunodeficiência, presença de infecções de repetição e doença reumática. A tabela 2 compreende antecedentes familiares, manifestações extra-articulares e exames complementares desses 11 pacientes.
Dos 11 pacientes com diagnóstico de imunodeficiência humoral (0,2%), Def IgA foi encontrada em oito, imunodeficiência comum variável em dois e deficiência de subclasses de IgG em um. A proporção entre sexo feminino e masculino foi de 7:4. A média da idade de início dos sintomas articulares e a média do diagnóstico da imunodeficiência foi de 6,2 anos (variou de um a 15) e 8,2 anos (variou de três a 19), respectivamente.
Cinco dos 11 pacientes descritos apresentaram artrite aguda com envolvimento de grandes articulações do esqueleto apendicular. A presença de oligoartrite aguda ocorreu nos três pacientes com LESJ (casos 6, 8 e 9), poliartrite aguda recorrente em um (caso 10) e oligoartrite aguda recorrente em outro (caso 2).
Seis pacientes apresentaram artrite crônica não-erosiva (AIJ símile), e um deles apresentava limitações articulares em coluna cervical, punhos, metacarpofalangeanas, joelho direito e tornozelos. Comprometimento oligoarticular foi observado em um paciente, e poliartrite foi observada em cinco. Estudo radiológico foi realizado em todos os indivíduos e nenhum deles apresentou erosões, cistos ósseos ou anquilose.
Um aspecto relevante deste estudo foi evidenciado em duas pacientes com LESJ que apresentaram deficiência seletiva de IgA associada a diabetes melito tipo I de difícil controle e necessidade de bomba de infusão contínua de insulina. Uma delas apresentou pneumonias de repetição associadas com uma doença auto-imune sistêmica com tireoidite, diarréia crônica, doença pulmonar obstrutiva crônica, vasculite de sistema nervoso central, com presença de múltiplos auto-anticorpos, tais como: antiperoxidase, antiinsulina e antigliadina IgG. Outra teve glomerulonefrite membranoproliferativa, tireoidite, com presença de múltiplos auto-anticorpos: TRAB, anti-GAD e antiinsulina.
DISCUSSÃO
O presente estudo evidencia baixa prevalência de imunodeficiência humoral primária em associação com artrite na população de pacientes atendidos em um serviço terciário de reumatologia pediátrica (0,2%).
A Def IgA é a imunodeficiência primária de maior prevalência(6,12,13). Neste estudo, oito dos 11 pacientes descritos apresentaram essa imunodeficiência humoral. Apesar de a etiologia da Def IgA ainda ser desconhecida, auto-anticorpos anti-IgA podem exercer um papel em sua indução(12,14). Petty et al.(5) evidenciaram uma freqüência de anticorpos anti-IgA entre 19% e 100% dos pacientes com essa imunodeficiência. Além disso, esses autores mostraram maior prevalência desses auto-anticorpos em pacientes com associação de Def IgA com AIJ ou LES (77% e 100%, respectivamente). Dessa forma, a Def IgA com anticorpos anti-IgA presentes poderia ser considerada um distúrbio auto-imune(14). Outro aspecto relevante é que a Def IgA secundária a drogas como antiinflamatórios não-hormonais, ouro parenteral e D-penicilamina é freqüentemente descrita(15-21). Nove dos onze pacientes avaliados neste estudo desenvolveram imunodeficiências previamente ao uso de drogas anti-reumáticas. O paciente 2 fez uso apenas intermitente e esporádico de antiinflamatórios não-hormonais durante os episódios de artrite aguda. O paciente 9 fez uso de prednisona, ciclofosfamida e micofenolato mofetil dos 15 aos 19 anos, porém seus níveis séricos de IgA nunca haviam sido realizados antes, o que impossibilita relacionar a Def IgA à terapêutica da doença auto-imune.
As doenças reumatológicas mais freqüentemente associadas com Def IgA são artrite reumatóide, AIJ e LESJ. São descritas mais raramente associações dessa imunodeficiência com esclerodermia, dermatopolimiosite e doença mista do tecido conectivo(2). No presente estudo, dos oito pacientes com Def IgA avaliados, quatro apresentaram artrite crônica e três apresentaram diagnóstico de LESJ. Outra paciente apresentou oligoartrite aguda e úlceras orais recorrentes, mas não preencheu critérios diagnósticos para LESJ.
De acordo com a literatura, o quadro clínico, a proporção entre os sexos e a idade de início da artrite não diferem em pacientes com Def IgA isolada ou associada à AIJ. A distribuição dos tipos de início da AIJ associada à Def IgA é: oligoarticular em 64%, poliarticular em 32% e sistêmica em 4% dos casos(22-24). Neste estudo, três pacientes com artrite AIJ símile apresentaram início e evolução poliarticular e um paciente apresentou início e evolução sistêmicos. Na maioria dos pacientes, o curso da doença é leve com poucas limitações funcionais e sem lesões radiográficas, conforme evidenciado nos pacientes deste trabalho.
Um dos pacientes com Def IgA apresentou, além de artrite crônica, eritema nodoso recorrente como primeira manifestação da imunodeficiência primária. Essa associação tem sido raramente descrita na literatura. Tremeau-Martinage et al.(25) descreveram um caso de um paciente de sete anos de idade com Def IgA, paniculite auto-imune e presença de múltiplos auto-anticorpos.
Doença auto-imune ocorre em 20% a 30% de pacientes com IDCV e a prevalência de artrite associada a essa imunodeficiência varia de 20% a 30%(2,26,27). Em uma série de 103 pacientes com IDCV, três pacientes apresentaram artrite crônica semelhante à AIJ(26). Pipitone et al.(8) relataram maior prevalência de AIJ, LES, síndrome de Sjögren e cirrose biliar primária em pacientes com IDCV. No presente estudo, os dois pacientes com IDCV apresentaram artrite crônica.
Em relação à deficiência de subclasses de IgG, há poucos relatos de associação com doenças auto-imunes. Aucouturier et al.(28) descreveram uma associação de deficiên-cia de IgG2 com vasculite e citopenias em uma grande série de 450 pacientes portadores dessa imunodeficiência humoral. O único paciente desse estudo com deficiência de IgG2 apresentou poliartrite aguda intermitente de caráter inespecífico, associada com entesite e diarréia crônica. O diagnóstico de doença inflamatória intestinal foi descartado pela colonoscopia e pela biópsia dirigida.
Embora as infecções sejam as manifestações mais comuns e mais precoces das imunodeficiências primárias, doen-ças auto-imunes também são freqüentes nesses pacientes. A associação entre imunodeficiências humorais primárias e doenças reumatológicas pode ser explicada por algumas hipóteses. Defeitos do sistema imune podem resultar em dificuldade de depuração de agentes microbianos, resultando ativação imunológica crônica e sintomas auto-imunes. Alelos de HLA que predispõem tanto para a auto-imunidade quanto para a imunodeficiência, como o HLA-B8, DR3 e Cw6, comuns à doença celíaca e Def IgA, também podem estar relacionados com a ocorrência dessa associação (29,30). Além disso, as manifestações reumatológicas dos pacientes com imunodeficiências primárias, em sua maioria, são inespecíficas e não são preenchidos os critérios diagnósticos para AIJ ou LESJ. Essas manifestações poderiam ser consideradas, portanto, uma conseqüência da imunodeficiência primária. Por sua vez, nos pacientes com doença auto-imune que preencheram critérios de classificação para diagnósticos de AIJ ou LESJ, é muito difícil determinar se as manifestações articulares estão relacionadas à própria colagenose ou à imunodeficiência humoral. Nesses casos, existe uma tendência a se considerar essa imunodeficiência um evento paralelo ao quadro articular. Entretanto, em ambos os casos (de doença auto-imune específica ou AIJ/LESJ símile), a freqüência de imunodeficiência humoral é maior do que na população geral. Dessa forma, a associação entre imunodeficiência e auto-imunidade em um mesmo paciente poderia representar um espectro clínico causado por uma única anormalidade imunológica. A maior compreensão das bases moleculares das imunodeficiências e de suas conseqüências para o sistema imune poderá contribuir para um melhor entendimento da fisiopatologia das doenças auto-imunes.
A presença de infecções de repetição e história familiar de imunodeficiência e doença auto-imune reforça a necessidade de pesquisa rotineira das imunoglobulinas séricas nos pacientes com artrite. Além disto, esses exames devem ser preferencialmente solicitados nos pacientes com LESJ e AIJ, particularmente com infecções de repetição e história familiar de doença auto-imune.
Recebido em 21/06/07.
Aprovado, após revisão, em 16/10/07.
Declaramos a inexistência de conflitos de interesse.
Unidades de Reumatologia, Alergia e Imunologia Pediátricas Instituto da Criança do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (ICr-HC-FMUSP).
-
1Wang LH, Tsai MJ, Huang MT, Lin SC, Chiang BL: Autoimmune manifestations in patients with primary immunodeficiency. Acta Paediatrics Taiwan 40: 243-9, 1999.
-
2Wulffraat NM, Sanders LA, Kuis W: Immunodeficiencies and rheumatic diseases. In: Cassidy JT, Petty RE. Textbook of Pediatric Rheumatology. W.B. Saunders Company, 4th edition, 2005. p. 706-23.
-
3Cassidy JT, Oldham G, Platts-Mills TAE: Functional assessment of a B-cell defect in patients with selective IgA deficiency. Clin Exp Immunol 35: 296-305, 1979.
-
4Pelkonen P, Savilahti E, Mäkelä AL: Persistent and transient IgA deficiency in juvenile rheumatoid arthritis. Scandinavian J Rheumatol 12: 273-9, 1983.
-
5Petty RE, Palmer NR, Cassidy JT, et al.: The association of autoimmune diseases and anti-IgA antibodies in patients with selective IgA deficiency. Clin Exp Immunol 37: 83-8, 1979.
-
6Liblau RS, Bach JF: Selective IgA deficiency and autoimmunity. Int Arch Allergy Immunology 99: 16-27, 1992.
-
7Cleland LG, Bell DA: The occurrence of systemic lupus erythematosus in two kindreds in association with selective IgA deficiency. J Rheumatol 5: 288-93, 1978.
-
8Pipitone N, Fioravanti A, Marcolongo R, Pitzalis C: Articular involvement in the course of primary hypogammaglobulinemia. Recenti Prog Med 92: 63-7, 2001.
-
9Bonilla FA, Bernstein IL, Khan DA, et al.: American Academy of Allergy, Asthma and Immunology; American College of Allergy, Asthma and Immunology; Joint Council of Allergy, Asthma and Immunology. Practice parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol 94: S1-63, 2005.
-
10Hochberg MC: Updating the American College of Rheuma-thology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 40: 1725, 1997.
-
11Petty RE, Southwood TR, Manners P, et al.: International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol 3: 390-2, 2004.
-
12Oen F, Petty RE, Schroeder ML: Immunoglobulin A deficiency: genetic studies. Tissue Antigens 19: 174-82, 1982.
-
13Cassidy JT, Nordby GL: Human serum immunoglobulin concentrations: prevalence of immunoglobulin deficiencies. J Allergy Clin Immunol 55: 35-48, 1975.
-
14Petty RE, Sherry DD, Johannson J: Anti-IgA antibodies in pregnancy. N Engl J Med 313: 1620-5, 1985.
-
15Barkley DO, Hohermuth HJ, Howard A, et al.: IgA deficiency in juvenile chronic arthritis. J Rheumatol 6: 219-24, 1979.
-
16Itescu S: Adult immunodeficiency and rheumatic disease. Rheum Dis Clin North Am 22: 53-72, 1996.
-
17Stanworth DR, Johns P, Williamson N, et al.: Drug-induced IgA deficiency in rheumatoid arthritis. Lancet 1: 1001-2, 1977.
-
18van Riel PL, van de Putte LB, Gribnau AJ, et al.: IgA deficiency during aurothioglucose treatment. Scand J Rheumatol 13: 334-6, 1984.
-
19Delamere JP, Farr M, Grindulis KA: Sulphasalazine induced selective IgA deficiency in rheumatoid arthritis. Br Med J 286: 1547-8, 1983.
-
20Savilahti E: Sulphasalazine induced immunodeficiency. Br Med J 287: 759, 1983.
-
21Leickly FE, Buckley RH: Development of IgA and IgG2 subclass deficiency after sulphasalazine therapy. J Pediatr 108: 481-2, 1986.
-
22Cassidy JT, Petty RE, Sullivan DB: Occurrence of selective IgA deficiency in children with juvenile rheumatoid arthritis. Arthritis Rheum 20: 181, 1977.
-
23Pelkonen P, Salilahti E, Westeren L, et al.: IgA deficiency in juvenile rheumatoid arthritis. Scand J Rheumatol 8: 4, 1975.
-
24Cassidy JT, Burt A, Petty RE, et al.: Prevalence of IgA deficiency in patients with connective tissue disease. N Engl J Med 280: 275, 1969.
-
25Tremeau-Martinage C, Bayle-Lebey L, Grouteau E, Bazex J: Localized lipoatrophy with persistent circulating autoantibodies and partial immunoglobulin A deficiency in a child. Dermatology 192: 353-7, 1996.
-
26Cunnibgham-Rundles C: Clinical and Immunological analyses of 103 patients with common variable immunodeficiency. J Clin Immunol 9: 22-3, 1989.
-
27Lee AH, Levinson AI, Schumacher HR: Hypogammaglobulinemia and rheumatic disease. Semin Arthritis Rheum 22: 252-64, 1993.
-
28Aucouturier P, Lacombe C, Bremard C, et al.: Serum IgG subclass levels in patients with primary immunodeficiency syndromes or abnormal susceptibility to infections. Clin Immunopathol 51: 22-37, 1989.
-
29Sleasman JW: The association between immunodeficiency and the development of autoimmune disease. Adv Dent Res 10: 57-61, 1996.
-
30Klemola T, Savilahti E, Koskimies S, Pelkonen P: HLA antigens in IgA deficient paediatric patients. Tissue Antigens 32: 218-23, 1988.
Datas de Publicação
-
Publicação nesta coleção
31 Jan 2008 -
Data do Fascículo
Dez 2007
Histórico
-
Aceito
16 Out 2007 -
Recebido
21 Jun 2007