
Abstract
The quantum mechanical problem posed by the internal motion of three particles subject to Coulomb interactions is variationally solved by means of an Eckart-Gaussian (EG) ansatz that exhibits an exponential behavior with respect to the radial coordinates {r1,r2}, and a harmonic Gaussian-type dependence on the interparticle distance r12, thereby providing explicit correlation. The proposed wavefunction is of the form (e-alpha1r1-beta1r2 + e-beta2r1-alpha2r2) r l12 e<FONT FACE="Symbol">-(g</FONT> 2r1-u0)2, through which ground state energies are calculated for a few two-electron atoms-considering finite nuclear mass effects-and molecular ions corresponding to electronic and mesonic systems. The physical interpretation and advantages of the EG wavefunction are discussed in terms of the relative masses of the particles in the analyzed systems. A useful application of the variational method is presented where the underlying structure of the 3-body wavefunction combines an atomic- and a molecular-like description of the system. The obtained energies agree with the exact results within 10-4 - 10-2 Hartrees.
Variational description of the 3-body Coulomb problem through a correlated Eckart-Gaussian wavefunction
A. Flores-Riveros and J. F. Rivas-Silva
Instituto de Física,
Benemérita Universidad Autónoma de Puebla,
Apartado Postal J-48, 72570 Puebla, Pue., Mexico
Received 26 May, 1999
The quantum mechanical problem posed by the internal motion of three particles subject to Coulomb interactions is variationally solved by means of an Eckart-Gaussian (EG) ansatz that exhibits an exponential behavior with respect to the radial coordinates {r1,r2}, and a harmonic Gaussian-type dependence on the interparticle distance r12, thereby providing explicit correlation. The proposed wavefunction is of the form (e-a1r1-b1r2 + e-b2r1 -a2r2) rl12 e-g( r12-u0)2, through which ground state energies are calculated for a few two-electron atoms-considering finite nuclear mass effects-and molecular ions corresponding to electronic and mesonic systems. The physical interpretation and advantages of the EG wavefunction are discussed in terms of the relative masses of the particles in the analyzed systems. A useful application of the variational method is presented where the underlying structure of the 3-body wavefunction combines an atomic- and a molecular-like description of the system. The obtained energies agree with the exact results within 10-4 - 10-2 Hartrees.
I Introduction
Three particles interacting via Coulomb forces represents a fundamental problem in quantum mechanics whose approximate solution provides some insight into the more complex analysis associated with few-body problems. Three-body Coulomb systems comprise a variety of diatomic molecular ions, e.g. hydrogenic and their isotopic species like H2+ [1], HD+, HT+ and DT+ [2], as well as exotic systems of interest in muon catalyzed fusion such as ppm+, ddm+, dtm+ and ttm+ [3]. Coulomb systems involving three particles relate also to the analysis of matter-antimatter coexistence, as that rendered by the experimental observation of antiprotonic helium ( He+) [4] and the formation of positronium ions Ps- (e-e-e+) through collisions of positron with atomic hydrogen [5] and other experimental techniques [6]. The study of two-electron atoms including the effect of finite nuclear mass to investigate bound [7] and resonant structure [8] involves the quantum mechanical analysis of three particles undergoing electrostatic interactions.
He+) [4] and the formation of positronium ions Ps- (e-e-e+) through collisions of positron with atomic hydrogen [5] and other experimental techniques [6]. The study of two-electron atoms including the effect of finite nuclear mass to investigate bound [7] and resonant structure [8] involves the quantum mechanical analysis of three particles undergoing electrostatic interactions.
H2+, being the simplest molecular species, has been the subject of numerous studies to illustrate the separation of electronic and nuclear motion as prescribed by the Born-Oppenheimer approximation [9], and it has been analyzed under adiabatic [10] and nonadiabatic treatments both within CI (Configuration Interaction) schemes [11] and by means of correlated wavefunctions variationally optimized [12]. In connection with the latter, attempting accurate descriptions of ground and excited state properties for 3-particle systems involves multiparameter set optimizations that often represent a challenging numerical task. Therefore, a suitable choice of the trial wavefunction and an efficient handling of the numerical optimization, are important aspects to consider.
The earliest attempts to describe two-electron systems in a nonadiabatic fashion, within the infinitely heavy nucleus approximation, led to propose trial wavefunctions of the form

where N is a normalization constant and a is a variational parameter. Correlation is thus introduced via a polynomial function P(r1,r2,r12) that depends on the interelectronic distance

as a third coordinate, in addition to the electron-nucleus distances r1 and r2. Ever since, Hylleraas coordinates,

have been widely used to expand wavefunctions variationally optimized in basis sets [13] that depend on these variables.
Three-particle systems for electronic (including finite-size corrections) [7a,7b] and mesomolecular species [14], have been described through correlated Slater-geminals

where coefficients Ckare found by solving the secular equation (Rayleigh-Ritz variational method) and the exponents ak, bk and gkare variationally optimized within selected samples of parameter sets. For each expansion the procedure involves optimization of six parameters to be varied on pseudorandom sequences.
Wavefunctions expanded in generalized Hylleraas basis sets [15]

have also been utilized for the variational description of 3-body Coulomb systems where optimization techniques similar to those of correlated Slater-geminals are followed.
With the above wavefunctions relatively low expansions are sufficient to accomplish reasonably accurate energies to describe ground and low excited states of electronic molecular ions. By contrast, obtaining the energy spectrum for mesomolecular systems (e.g. ddm+, dtm+ and ttm+), where m is the binding particle, becomes a remarkably more difficult task. This is essentially due to the presence of the large muon mass, outweighing that of electrons by more than two orders of magnitude (mm = 207me), which makes these systems strongly nonadiabatic. Indeed, mesomolecular species, which play a key role in muon catalyzed fusion processes, consist of two isotopic hydrogen nuclei and a muon, all three tightly bound via Coulomb interactions, where high vibrational energies overcome the electrostatic repulsion driving the nuclei at so short distances from each other-in fact, within the strong forces interaction range-that fusion eventually occurs. From a theoretical point of view, these molecular ions were originally analyzed through wavefunctions expressed in adiabatic expansions [16], and later nonadiabatically approached by means of correlated basis sets [15]. In either treatment, considerably large expansions are necessary to reach convergence within an accuracy of fractions of millielectronvolts. It would thus be desirable to construct a wavefunction giving a high accuracy with a reduced expansion length. As a first step to this end, we here propose an ansatz that combines an atomic- and a molecular-like character of the form

where a1 = pa, b1 = qb, a2 = qa, b2 = pb, and a, b, g and u0 are variational parameters, whereas p and q are asymmetry factors defined as

that depend on the masses of particles 1 and 2, whose distances to particle 3 are denoted by r1 and r2, respectively. Hence, for homonuclear systems (m1=m2; p=q=1) the above wavefunction is symmetric under exchange of these coordinates, i.e. YEG (r1,r2) = YEG (r2,r1), whereas for heteronuclear systems (m1 > m2), it is asymmetric: YEG (r1,r2) ¹ YEG (r2,r1). (Note that the operation r1
r2 is not to be applied on the labels of masses m1 and m2, because this would lead to a symmetric combination for either homonuclear or heteronuclear systems.)
The atomic-like character of YEG is clearly associated with the symmetric (or asymmetric) combination of exponential functions, whereas the Gaussian factor r12l e-g(r12 - u0)2, denotes a harmonic oscillator-type function that describes vibrational motion on the interparticle coordinate r12, around an equilibrium distance u0. The first part of the wavefunction,

corresponds to a generalized Eckart function [17], which was the earliest variational uncorrelated ansatz (symmetric combination) utilized to describe two-electron atoms, assuming an infinitely heavy nuclear mass, where through separate screening factors on each coordinate a more flexible description of the atom is attained, as compared to the simpler function

In order to assess the accuracy of the here proposed Eckart-Gaussian wavefunction, YEG, we set out to make a systematic comparison with that obtained through a generalized Hylleraas function,

which is the most similar to the former that has been utilized in a correlated description of 3-body Coulomb systems. (The set {a1,b1,a2,b2} relates to variational parameters a and b via asymmetry mass-dependent factors in the same fashion as given for the EG function.) In this report we compare variational ground state energies as obtained for the Eckart-Gaussian and Generalized Hylleraas ansätze, Eqs. (6) and (10), respectively, for a variety of atomic and molecular species, comprising three charged particles subject to electrostatic interactions.
II Theory
We consider the nonrelativistic 3-body Coulomb Hamiltonian,

where the mij's refer to reduced masses of particles i and j (it is assumed that m1³ m2), m3 denotes the mass of particle 3 and z1,z2,z3 are the charges of particles 1, 2 and 3, respectively. The masses involved in the analyzed systems are given in atomic units: me = 1.0, mm = 206.7686, mp = 1836.1515, md = 3670.481, mt = 5496.899 and ma = 7294.295, corresponding to the electron, proton, deuteron, triton and a (helium nucleus) particles, respectively. The mass-polarization term gives the finite nuclear mass correction (proportional to the scalar product of  1 and 2), which for an atomic hamiltonian is absent in the infinitely heavy nucleus approximation (m3® ¥).
1 and 2), which for an atomic hamiltonian is absent in the infinitely heavy nucleus approximation (m3® ¥).
When expressed in Hylleraas coordinates the above Hamiltonian reads,
 (12)
(12)
where
1,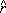 2 and 12 denote the unit vectors for distances between particles 1-3, 2-3 and 1-2, respectively.
2 and 12 denote the unit vectors for distances between particles 1-3, 2-3 and 1-2, respectively.
The expectation value of the Hamiltonian with respect to the EG and GH trial functions,

leads to integrals of the form

where the differential volume is
dV = 8p2r1r2r12dr1dr2dr12 (15)
The above integral can be expressed in terms of Hylleraas coordinates s, t and u:
0 £ s £ ¥; 0 £ u £ s; -u £ t £ u, (16)
which relate to r1, r2 and r12 as

We thus obtain,

which leads to

It is straightforward to prove than an integral of the form

can be written as

for B ¹ 0, and

for B = 0.
GNML (and therefore FNML) can analytically be evaluated for C2 = 0 (GH functions). Otherwise (EG functions), one deals with the improper integrals

which can be calculated through the recursion formula
2p2IN+1 + p1IN - N IN-1 = 0; N ³ 1 (24)
The ones of lowest order are given by

where erfc (z) denotes the complementary error function,

which can be calculated for any argument with standard numerical methods.
III Results and Discussion
Energies for EG and GH trial wavefunctions were varied with respect to the four nonlinear {a,b,g,u0} and three nonlinear parameters {a,b,c}, respectively, by means of an algorithm based on a numerical quasi-Newton method to minimize a multivariable function. Optimization for either case took a negligibly short processing time. Converged optimal values were attained within a gradient magnitude in the range 10-6-10-5, for which analytical expressions for the energy gradients were also calculated.
For the sake of simplicity, powers {n,m,l} for the corresponding coordinates r1, r2 and r12, in both trial functions, were chosen so as to fulfill the condition n+m+l £ 2. Throughout all calculated systems, the optimal powers, giving the lowest variational energies for the two wavefunctions commensurate with this condition, were found to consistently span the set n=m=0 and l either equal to 0, 1 or 2. Thus, the actual ansätze that we here investigate are of the form
YEG = (e-a1r1-b1r2 + e-b2r1 -a2r2) rl12 e-(g 2r1-u0)2, (27)
(as stated in the abstract of this report) and
YGH = (e-a1r1-b1r2 + e-b2r1 -a2r2) rl12 e-c r12, (28)
In Table 1 variationally optimized ground state energies are given for 3-body Coulomb systems classified-for interpretive purposes-in three classes: two-electron atoms, molecular electronic ions and mesomolecular species.
In descending order, the three frames contains data for two-electron atons, molecular eletronic ions and mesomolecular species, respectively. Optimal energy (E) amd energy difference (E - Eex) coresponding to EG and GH trial functions are given on the first and second row, respectively, for each entry.

As seen in this table, the EG function consistently yields lower variational energies than those obtained with function GH, except for the muonic molecule ppm+. It is interesting to note that for the two-electron atoms here analyzed the molecular description gives a better result. In this case, the vibrational part of function EG describes the electronic motion within the Coulomb field created by the binding particle, which is a positively charged nucleus. In a way, this is the opposite view of the Born-Oppenheimer approximation, where the fast motion of the light particles among themselves proceeds in the presence of a heavy body, although in a nonadiabatic fashion since no distance is ever fixed to solve the Schrödinger equation. However, the optimal energies obtained for the two functions are very similar, thus indicating that the intrinsically atomic part of the EG wavefunction provides an essential contribution, where by comparison the vibrational ansatz barely improves the overall description. Surprisingly, this trend shows up even for the lightest atomic ion, Ps- (e-e-e+), where the three particles possess the same mass and the ground state is very diffuse, albeit in this case we obtain the most alike optimal energies for the two functions, i.e. we deal with the most atomic-like molecule of all.
As also shown in the same table, ground state energies for electronic molecular ions, as obtained with function EG, are the most accurate of all homonuclear systems here analyzed-including the exotic molecular species Mu2+ (m+m+e-)-where the slow motion of the heavy particles in the presence of an electron is adequately described via the molecular ansatz. In contrast to the previous case, the atomic part of the wavefunction plays a less significant role to improve the global description of these molecular species, as inferred from the energies optimized through function GH, which remain in average .03 Hartrees off the exact values, as compared to the substantially more accurate energies that the EG wavefunction yields (being 3 ×10-4 to 10-3 Hartrees above the exact nonrelativistic values). In fact, for molecules with increasingly heavier nuclei a higher accuracy in the energies is accomplished. Accordingly, description of vibrational internuclear motion correspondingly becomes a more important feature. Kolos et al. [12a] report a ground state energy of -0.58305 Hartrees for H2+ through a 32-term GH expansion, which compared to that obtained with our one-term EG function, -0.59643 Hartrees, clearly indicates the latter's higher variational accuracy.
Mesomolecular species present the most challenging systems upon variational analysis. These molecules are formed under extreme conditions of chemical confinement, comprising three massive particles tightly bound, where atomic and molecular characters are strongly intermingled. Their accurate description thus calls for an approach where both features are explicitly accounted for. The optimal energies obtained through functions EG and GH, as seen in Table 1, clearly show that a purely atomic description, despite the built-in correlation in the wavefunction, does not lead to a higher variational accuracy. With the exception of ppm+, the EG function yields a lower energy for homonuclear as well as heteronuclear molecules, especially so for the heaviest species ddm+, dtm+ and ttm+. Note that for these three the energies obtained with both trial functions differ to a greater extent than those optimized for the two-electron atoms. This is consistent with our conjecture: the vibrational ansatz plays a more important role in highly confined Coulomb systems, like mesomolecules. It is interesting to note that the one case where the GH function gives a marginally lower energy, ppm+, corresponds to the mesonic molecule where nuclei and binding particle have the least unequal masses (m3/m1 = 0.11).
For heteronuclear systems the present findings show a lesser degree of accuracy, the EG function being nevertheless variationally superior. The description of three different particles becomes a more difficult task, where one no longer has the advantage of using a symmetric wavefunction. These systems are usually described through unsymmetrized wavefunctions, i.e. we could have chosen trial functions of the form

and

instead of the here proposed EG and GH wavefunctions, respectively. However, at the present variational level where only one-term functions are analyzed, the above ansätze were found-in the course of test calculations-to give remarkably poor energies upon optimization. The asymmetry we introduce in the wavefunctions is more advantageous since it preserves the same structure for symmetric and asymmetric combinations, and depends on the particle masses of the specific heteronuclear system under study. The latter is thus physically described in a more meaningful way since the degree of asymmetry is dictated by its particular characteristics.
The optimal parameters for the EG and GH wavefunctions, associated with true minima corresponding to vanishing energy gradient (|
E| £ 10-5), are given in Table 2. In general, the pair of linear exponents for each function, {a,b} and {a,b}, respectively, are very similar, which is to be expected since they correspond to the same Eckart-type function for either variational ansatz. For most of two-electron atoms optimal parameter c is negative and thus factor e-cr12 yields a sizable contribution, which physically reflects the fact of having two fermions with paired spins (thus spin-uncorrelated) that spatially become highly correlated in the 1s-orbital. This feature is apparent even for positronium, Ps-, though in a less pronounced fashion since the corresponding optimal parameter c is in this case positive and factor e-cr12 is thus smaller in magnitude. This is consistent with a highly diffuse ground state that this atom is known to possess, i.e. the electrons are far less confined in the 1s-orbital within the positron Coulomb field than in the presence of a heavy nucleus. EG function's optimal parameter g is fairly small for two-electron atoms where through a minimal contribution of the Gaussian factor e-g r122 (g << 1 Þ e-g r122~ 1), the ansatz allows for a prevalence of atomic character, leading thereof to their most suitable representation. By contrast, this factor becomes substantially more important for the electronic molecular ions where a large optimal parameter g points to an enhanced vibrational and localized character, essential for the appropriate description of nuclear motion in these systems. For mesomolecular species, optimal parameter g becomes smaller by comparison, though not as small as in the case of two-electron atoms, where the EG function yields atomic and molecular character to roughly the same extent. This is in line with the interpretation above discussed regarding the physical features of mesonic molecules.First and second row on each entry contain the optimal power of r12 (l) and optimized nonlinear parameters for YEG and YGH functions, respectively.
Optimal parameters for EG and GH trial functions as obtained for 3-body Coulomb systems.
In order to further test the accuracy of the here proposed EG trial function, we have performed a series of calculations for all systems considered in Table 1 by using 4- and 10-term Hylleraas trial functions, expressed in coordinates s, t and u, which represent classical ansätze for the variational description of two-electron atom ground states, proposed by Hylleraas in his early work [18]. We point out that the calculations here performed were obtained by using the full 3-body Coulomb Hamiltonian (see Eq. (11)), and not within the infinitely heavy nucleus approximation (adopted in Hylleraas' approach).
In Table 3 it is shown the accuracy of our EG function for two-electron atoms and molecular ions in comparison to that of the 4- and 10-term Hylleraas functions,

for homonuclear systems, and

for heteronuclear species. The expansions for the former are symmetric under exchange of coordinates r1 and r2, i.e. s ® s, t ® -t and u ® u, thus, s, u and t2 are invariant under this operation and YS (s,t,u) = YS (s,-t,u), whereas those for the latter are correspondingly asymmetric, i.e. terms containing t reverse sign and therefore YA(s,t,u) ¹ YA(s,-t,u). These particular expansions were chosen because, firstly, the 4-term function contains the same number of variational parameters (h, c1, C2 and c3) as our one-term EG trial function, which is spanned by four nonlinear parameters, and their variational ability can thus be compared on fair grounds (notice also that both trial functions contain the correlation factor u = r12). Secondly, the inclusion of 10 terms should, to a reasonable extent, provide information on the variational ability of a given basis set for a minimal expansion. The variational energies here given were fully optimized and correspond to an energy gradient magnitude averaging ~ 10-6 throughout, for which analytical expressions of the gradient were used.
Optimized energies for atomic and molecular species. Energies E4 and E10 correspond to those obtained with YS4,A4 and YS10,A10 Hylleraas trial functions, whereas EEG refers to that of EG trial function's (as given in Table 1). Energy differences (E-Eex) are given on the second row for each entry.
Ground state energies for 3-body Coulomb systems optimized with 4- and 10-term Hylleraas functions as well as EG trial function.
From these results it is clear that the atomic systems are unfavorably described by our one-term EG function in comparison to the Hylleraas expansions (although it es marginally more accurate than function YS4 for Mu-, H-, D- and T-), which is not surprising since the EG function's vibrational part (e-g(r12-u0)2) becomes inadequate and unphysical when associated with the lightest particles of the system (r12 corresponds to interelectronic distance), in the presence of a third massive body (nucleus).
By contrast, the feature that for two-electron atoms is unphysical becomes precisely what is desirable in molecular ions since the vibrational part of function EG is in this case associated with the nuclear motion (r12 corresponds to internuclear distance), which is also in line with the fundamental concept on which the Born-Oppenheimer approximation is based: The relatively slow motion of the nuclei proceeds favorably in the presence of a light binding particle (electron or muon). Results in Table 3 unquestionably show that the electronic molecules and mesomolecular species here analyzed are far better described by the one-term EG function than by any of the Hylleraas expansions.
In this report, rather than describing two-electron atoms, we are aimed at improving a variational description for electronic and muonic molecular species, most especially the latter, which are known to require large correlated expansions to achieve a reasonable accuracy. We believe that the calculations here presented indicate that, for the latter systems, EG expansions are likely to attain a high variational accuracy upon relatively low expansions, at least lower than those that basis sets expressed in Hylleraas or GH-type trial functions would probably need.
IV Concluding Remarks
A trial wavefunction combining atomic and molecular character via an Eckart-type function times a correlated Gaussian ansatz has been proposed for the variational description of 3-body Coulomb systems. We have demonstrated the accuracy of this function by performing a systematic comparison with that obtained through a generalized Hylleraas basis consisting of one term as well as 4-term and 10-term Hylleraas functions. When applied on different atomic and molecular species, their variational precision is shown to be dependent on the relative masses between the binding particle and the the other two connected via interparticle distance r12, where in general the EG wavefunction is found to be more accurate for molecular systems.
The full variational ability of these functions must of course be established through a convergence analysis upon systematic increase of basis set expansions, performed e.g. via pseudorandom sequences spanned on variationally optimized intervals for the nonlinear parameters. Such intervals can be generated through random tempering formulas for selected low-order EG expansions. The latter deliver core functions over which a systematic increase of basis sets is performed, while keeping the nonlinear parameters fixed. This procedure is quite feasible and has previously been applied in the optimization of Slater-geminals, as mentioned in the introductory section. One of the present authors has recently utilized a similar procedure to optimize generalized Hylleraas-Gaussian basis sets applied to the variational description of two-electron atoms (within the infinitely heavy nucleus approximation) [19].
A convergence analysis for Eckart-Gaussian basis sets to describe bound structure of mesomolecular systems will be the subject of future investigation.
Acknowledgments
Financial support provided by Sistema Nacional de Investigadores (SNI) and partial provision of funds by Consejo Nacional de Ciencia y Tecnología (CONACYT) under project 1349 P-E, are gratefully acknowledged.
References
1. (a) S. Cohen, J. R. Hiskes and R. J. Riddell Jr., Phys. Rev. 119, 1025 (1960); (b) C. L. Beckel, M. Shafi and J. M. Peek, J. Chem. Phys. 59, 5288 (1973); (c) M. Shafi and C. L. Beckel, J. Chem. Phys. 59, 5294 (1973); (d) N. J. Kirchner, A. O'Keefe, J. R. Gilbert and M. T. Bowers, Phys. Rev. Lett. 52, 26 (1984).
2. (a) D. M. Bishop, Phys. Rev. Lett. 37, 484 (1976); (b) D. M. Bishop and R. W. Wetmore, Mol. Phys. 26, 145 (1973).
3. (a) W. H. Breunlich, P. Kammel, J. S. Cohen and M. Leon, Ann. Rev. Nucl. Part. Sci. 39, 311 (1989); (b) J. Rafelski and H. E. Rafelski, Particle World 2, 21 (1991); (c) L. Ponomarev, Contemp. Phys. 31, 219 (1991); (d) P. Froelich, Adv. Phys. 41, 405 (1992).
4. M. Iwasaki et al., Phys. Rev. Lett. 67, 1246 (1991).
5. W. Sperber et al., Phys. Rev. Lett. 68, 3690 (1992).
6. A. P. Mills Jr., Phys. Rev. Lett. 46, 717 (1981).
7. (a) A. M. Frolov and D. M. Bishop, Phys. Rev. A 45, 6236 (1992); (b) A. M. Frolov and V. H. Smith Jr., Phys. Rev. A 49, 3580 (1994); (c) M. I. Haftel and V. B. Mandelzweig, Phys. Rev. A 49, 3344 (1994).
8. Y. K. Ho, Phys. Rev. A 19, 2347 (1979).
9. F. R. Pilar, Elementary Quantum Chemistry (2nd edition, McGraw-Hill, 1990).
10. (a) D. R. Bates, K. Ledsham and A. L. Stewart, Phil. Trans. Roy. Soc. (London) A246, 215 (1954); (b) H. Wind, J. Chem. Phys. 43, 2956 (1965); (c) C. L. Beckel, B. D. Hansen III and J. M. Peek, J. Chem. Phys. 53, 3681 (1970).
11. (a) G. Blanke and H. Kleindienst, Int. J. Quant. Chem. 51, 3 (1994); (b) D. M. Bishop and L. M. Cheung, Int. J. Quant. Chem. 15, 517 (1979).
12. (a) W. Kolos, C. C. J. Roothaan and R. A. Sack, Rev. Mod. Phys. 32, 178 (1960); (b) A. Fröman and J. L. Kinsey, Phys. Rev. 123, 2077 (1961); (c) D. A. Kohl and E. J. Shipsey, J. Chem. Phys. 84, 2707 (1986).
13. (a) C. L. Pekeris, Phys. Rev. 112, 1649 (1958);
(b) C. L. Pekeris, Phys. Rev. 115, 1216 (1959);
(c) T. Kinoshita, Phys. Rev. 105, 1490 (1957).
14. (a) P. Petelenz and V. H. Smith Jr., Phys. Rev. A 36, 4078 (1987); (b) S. A. Alexander and H. J. Monkhorst, Phys. Rev. A 38, 26 (1988).
15. (a) K. Szalewicz, H. J. Monkhorst, W. Kolos and A. Scrinzi, Phys. Rev. A 36, 5494 (1987); (b) P. Froelich and A. Flores-Riveros, Phys. Rev. Lett. 70, 1595 (1993).
16. I. Vinitsky and L. I. Ponomarev, Sov. J. Part. Nucl. 13, 557 (1982).
17. C. Eckart, Phys. Rev. 36, 878 (1930).
18. (a) E. A. Hylleraas, Z. Physik 48, 469 (1928); (b) E. A. Hylleraas, Z. Physik 54, 347 (1929); (c) E. A. Hylleraas, Z. Physik 60, 624 (1930).
19. A. Flores-Riveros, Int. J. Quant. Chem. 66, 287 (1998).
20. K. P. Huber and G. Herzberg, Constants of Diatomic Molecules (Van Nostrand-Reinhold, New York, 1979).
- 1. (a) S. Cohen, J. R. Hiskes and R. J. Riddell Jr., Phys. Rev. 119, 1025 (1960);
- (b) C. L. Beckel, M. Shafi and J. M. Peek, J. Chem. Phys. 59, 5288 (1973); (c) M. Shafi and C. L. Beckel, J. Chem. Phys. 59, 5294 (1973);
- (d) N. J. Kirchner, A. O'Keefe, J. R. Gilbert and M. T. Bowers, Phys. Rev. Lett. 52, 26 (1984).
- 2. (a) D. M. Bishop, Phys. Rev. Lett. 37, 484 (1976);
- (b) D. M. Bishop and R. W. Wetmore, Mol. Phys. 26, 145 (1973).
- 3. (a) W. H. Breunlich, P. Kammel, J. S. Cohen and M. Leon, Ann. Rev. Nucl. Part. Sci. 39, 311 (1989);
- (b) J. Rafelski and H. E. Rafelski, Particle World 2, 21 (1991); (c) L. Ponomarev, Contemp. Phys. 31, 219 (1991);
- 4. M. Iwasaki et al., Phys. Rev. Lett. 67, 1246 (1991).
- 5. W. Sperber et al., Phys. Rev. Lett. 68, 3690 (1992).
- 6. A. P. Mills Jr., Phys. Rev. Lett. 46, 717 (1981).
- 7. (a) A. M. Frolov and D. M. Bishop, Phys. Rev. A 45, 6236 (1992);
- (b) A. M. Frolov and V. H. Smith Jr., Phys. Rev. A 49, 3580 (1994);
- (c) M. I. Haftel and V. B. Mandelzweig, Phys. Rev. A 49, 3344 (1994).
- 8. Y. K. Ho, Phys. Rev. A 19, 2347 (1979).
- 9. F. R. Pilar, Elementary Quantum Chemistry (2nd edition, McGraw-Hill, 1990).
- 10. (a) D. R. Bates, K. Ledsham and A. L. Stewart, Phil. Trans. Roy. Soc. (London) A246, 215 (1954);
- (b) H. Wind, J. Chem. Phys. 43, 2956 (1965); (c) C. L. Beckel, B. D. Hansen III and J. M. Peek, J. Chem. Phys. 53, 3681 (1970).
- 11. (a) G. Blanke and H. Kleindienst, Int. J. Quant. Chem. 51, 3 (1994);
- (b) D. M. Bishop and L. M. Cheung, Int. J. Quant. Chem. 15, 517 (1979).
- 12. (a) W. Kolos, C. C. J. Roothaan and R. A. Sack, Rev. Mod. Phys. 32, 178 (1960);
- (b) A. Fröman and J. L. Kinsey, Phys. Rev. 123, 2077 (1961); (c) D. A. Kohl and E. J. Shipsey, J. Chem. Phys. 84, 2707 (1986).
- 13. (a) C. L. Pekeris, Phys. Rev. 112, 1649 (1958);
- (b) C. L. Pekeris, Phys. Rev. 115, 1216 (1959);
- (c) T. Kinoshita, Phys. Rev. 105, 1490 (1957).
- 14. (a) P. Petelenz and V. H. Smith Jr., Phys. Rev. A 36, 4078 (1987);
- (b) S. A. Alexander and H. J. Monkhorst, Phys. Rev. A 38, 26 (1988).
- 15. (a) K. Szalewicz, H. J. Monkhorst, W. Kolos and A. Scrinzi, Phys. Rev. A 36, 5494 (1987);
- 16. I. Vinitsky and L. I. Ponomarev, Sov. J. Part. Nucl. 13, 557 (1982).
- 17. C. Eckart, Phys. Rev. 36, 878 (1930).
- 18. (a) E. A. Hylleraas, Z. Physik 48, 469 (1928);
- (b) E. A. Hylleraas, Z. Physik 54, 347 (1929);
- 19. A. Flores-Riveros, Int. J. Quant. Chem. 66, 287 (1998).
- 20. K. P. Huber and G. Herzberg, Constants of Diatomic Molecules (Van Nostrand-Reinhold, New York, 1979).
Publication Dates
-
Publication in this collection
07 Aug 2000 -
Date of issue
Sept 1999
History
-
Received
26 May 1999




