Abstracts
Catalytic promiscuity has emerged as an important property of many enzymes since the relationship of this property to enzymatic evolution became clear. Simultaneously, the development of suitable biomimetic catalytic systems capable of mimicking the promiscuous catalytic properties of such enzymes represents a new challenge for bioinorganic chemists. In this paper we report on the X-ray structure, the solution studies and the promiscuous catalytic activity of the mixed-valence complex [(bpbpmp)FeIII(µ-OAc)2FeII](ClO4), (1), containing the unsymmetrical dinucleating ligand 2-{[(2-hydroxybenzyl)(2-pyridylmethyl)aminomethyl]-4-methyl-6-[bis(2-pyridylmethyl)aminomethyl]}phenol (H2bpbpmp). Potentiometric and spectrophotometric titrations and kinetics studies showed that this coordination compound generates active species that promote hydrolytic cleavage of double strand DNA (dsDNA), with a rate enhancement of 1.9×10(8) over the non-catalyzed reaction, as well as promote oxidation of 3,5-di-tert-butylcatechol (3,5-dtbc), with k cat = 1.16 × 10-2 s-1 and K M = 7.1×10-4 mol L-1. Thus, complex 1 shows both hydrolase and oxidoreductase activities and can be regarded as a man-made model for studying catalytic promiscuity.
mixed-valence FeIII FeII complex; unsymmetrical ligand; catalytic promiscuity; diester and DNA cleavage; catecholase activity
Os estudos sobre enzimas têm mostrado que existe uma relação clara entre a evolução enzimática e a propriedade de promiscuidade catalítica que elas podem apresentar. Com isto, o desenvolvimento de sistemas biomiméticos capazes de atuar de modo promíscuo, de forma similar a algumas enzimas, mostrou-se um novo desafio para os químicos bioinorgânicos. Neste trabalho, reportamos a estrutura de raios X e os estudos em solução e de promiscuidade catalítica do complexo [(bpbpmp)FeIII(µ-OAc)2FeII](ClO4), (1), contendo o ligante binucleante não-simétrico 2-{[(2-hidroxibenzil)(2-piridilmetil)aminometil]-4-metil-6-[bis(2-piridilmetil)aminometil]}fenol (H2bpbpmp). Estudos de titulações potenciométrica e espectrofotométrica e cinéticos mostraram que esse composto de coordenação gera espécies ativas capazes de clivar o dsDNA (DNA de fita dupla) hidroliticamente, com um aumento de 1,9×10(8) na taxa de reação em comparação com a reação não catalisada, e capazes de oxidar o 3,5-di-terc-butilcatecol (3,5-dtbc), com k cat = 1,16 × 10-2 s-1 e K M = 7,1×10-4 mol L-1. Assim, o complexo 1 apresenta atividades de hidrolase e oxidoredutase, sendo um interessante modelo sintético para estudos de promiscuidade catalítica.
ARTICLE
Catalytic promiscuity: catecholase-like activity and hydrolytic DNA cleavage promoted by a mixed-valence FeIIIFeII complex
Ademir Neves* * e-mail: ademir@qmc.ufsc.br ,I; Adailton J. BortoluzziI; Rafael JovitoI; Rosely A. PeraltaI; Bernardo de SouzaI; Bruno SzpoganiczI; Antônio C. JoussefI; Hernán TerenziII; Patricia C. SeverinoII; Franciele L. FischerII; Gerhard SchenkIII; Mark J. RileyIII; Sarah J. SmithIII; Lawrence R. GahanIII
ILABINC, Departamento de Química
IICentro de Biologia Molecular Estrutural, Departamento de Bioquímica, Universidade Federal de Santa Catarina, 88040-900 Florianópolis-SC, Brazil
IIISchool of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane 4072, Australia
ABSTRACT
Catalytic promiscuity has emerged as an important property of many enzymes since the relationship of this property to enzymatic evolution became clear. Simultaneously, the development of suitable biomimetic catalytic systems capable of mimicking the promiscuous catalytic properties of such enzymes represents a new challenge for bioinorganic chemists. In this paper we report on the X-ray structure, the solution studies and the promiscuous catalytic activity of the mixed-valence complex [(bpbpmp)FeIII(µ-OAc)2FeII](ClO4), (1), containing the unsymmetrical dinucleating ligand 2-{[(2-hydroxybenzyl)(2-pyridylmethyl)aminomethyl]-4-methyl-6-[bis(2-pyridylmethyl)aminomethyl]}phenol (H2bpbpmp). Potentiometric and spectrophotometric titrations and kinetics studies showed that this coordination compound generates active species that promote hydrolytic cleavage of double strand DNA (dsDNA), with a rate enhancement of 1.9×108 over the non-catalyzed reaction, as well as promote oxidation of 3,5-di-tert-butylcatechol (3,5-dtbc), with kcat = 1.16 × 10-2 s-1 and KM = 7.1×10-4 mol L-1. Thus, complex 1 shows both hydrolase and oxidoreductase activities and can be regarded as a man-made model for studying catalytic promiscuity.
Keywords: mixed-valence FeIIIFeII complex, unsymmetrical ligand, catalytic promiscuity, diester and DNA cleavage, catecholase activity
RESUMO
Os estudos sobre enzimas têm mostrado que existe uma relação clara entre a evolução enzimática e a propriedade de promiscuidade catalítica que elas podem apresentar. Com isto, o desenvolvimento de sistemas biomiméticos capazes de atuar de modo promíscuo, de forma similar a algumas enzimas, mostrou-se um novo desafio para os químicos bioinorgânicos. Neste trabalho, reportamos a estrutura de raios X e os estudos em solução e de promiscuidade catalítica do complexo [(bpbpmp)FeIII(µ-OAc)2FeII](ClO4), (1), contendo o ligante binucleante não-simétrico 2-{[(2-hidroxibenzil)(2-piridilmetil)aminometil]-4-metil-6-[bis(2-piridilmetil)aminometil]}fenol (H2bpbpmp). Estudos de titulações potenciométrica e espectrofotométrica e cinéticos mostraram que esse composto de coordenação gera espécies ativas capazes de clivar o dsDNA (DNA de fita dupla) hidroliticamente, com um aumento de 1,9×108 na taxa de reação em comparação com a reação não catalisada, e capazes de oxidar o 3,5-di-terc-butilcatecol (3,5-dtbc), com kcat = 1,16 × 10-2 s-1 e KM = 7,1×10-4 mol L-1. Assim, o complexo 1 apresenta atividades de hidrolase e oxidoredutase, sendo um interessante modelo sintético para estudos de promiscuidade catalítica.
Introduction
Catalytic promiscuity means, in the broadest sense, the ability of a given natural or non-natural active site to catalyze more than one chemical transformation. It constitutes a very important property of many enzymes, having a natural role in evolution and, occasionally, in the biosynthesis of secondary metabolites.1-7 For example, it has been demonstrated that vanadium-dependent chloroperoxidase shows phosphatase activity when vanadate is not present, indicating that its active site is very similar to that of the acid phosphatases.8 On the other hand, it was found that an acid phosphatase from plants (in contrast to the kidney bean purple acid phosphatase (PAP)) uniquely exhibits chloroperoxidase activity with loss of phosphatase activity when orthovanadate is added to the apo form of the enzyme.9 Although these activities constitute very different overall reactions, they involve enzyme-bound species that are structurally related. Furthermore, it has been demonstrated that the PAP isolated from tomato may act as a bifunctional enzyme that operates in the hydrolysis of phosphate-esters and also displays significant alkaline peroxidase activity with the involvement of reactive oxygen species.10 Indeed, several plant PAPs have been reported to be expressed in response to pathogen attack or elicitor treatment.11,12 Nevertheless, no catecholase activity for any of the well characterized PAPs has as yet been described. While several enzymes displaying catalytic promiscuity have been the subject of recent related investigations,1-7 there are few examples of synthetic analogues which exhibit such multifunctional activities.13-18
In recent years, considerable attention has been devoted to the synthesis, X-ray structural analysis and physicochemical characterization of structural and functional dinuclear FeIIIMII mimetics for the active site of metallohydrolases such as purple acid phosphatase.19-21 In particular, we have successfully developed a general method for the preparation of mixed-valence homo- and hetero-dinuclear MIIIMII complexes, using the unsymmetrical ligand (H2bpbpmp) 2-{[(2-hydroxybenzyl)(2-pyridylmethyl)aminomethyl]-4-methyl-6-[bis(2-pyridylmethyl)aminomethyl]}phenol (Scheme 1).22-32
For example, it has been established that the [(bpbpmp)FeIII(µ-OAc)2FeII](ClO4)H2O complex, (1), is a good model for the electrochemical properties of uteroferrin, with a reduction potential for the FeIIIFeIII to FeIIIFeII couple of 380 mV (versus the normal hydrogen electrode, NHE), which compares well with the 344 mV measured for uteroferrin at pH 4.1 via direct electrochemistry and 367 mV at pH 5 via microcoulometry.9, 33,34 It has also been well documented that complex 1 and its corresponding oxidized complex [(bpbpmp)FeIII(µ-OAc)2FeIII](ClO4)2, (2), are good models for the resonance Raman properties of mammalian PAPs in their reduced and oxidized forms, respectively.30 In addition, to understand the spectral signatures of the dinuclear center and to relate these to the di-iron enzyme uteroferrin, spectroelectrochemical, spectroscopic, and computational studies, and phosphatase-like reactivity of the [(bpbpmp)FeIII(µ-OAc)2FeII]+ ion complex have also been reported.31 Very recently, the [(bpbpmp)FeIII(µ-OAc)2FeII]+ complex has been used as an electrochemical biomimetic sensor for the determination of phenolic compounds.32
In this study, we describe the X-ray structure and solution studies of the mixed-valence complex [(bpbpmp)FeIII(µ-OAc)2FeII](ClO4)H2O, (1), which is able to cleave phosphodiester bonds of the activated substrate bis(2,4-dinitrophenyl)phosphate (2,4-bdnpp)35 and of DNA itself, but also shows significant catalytic activity in the oxidation of the model substrate 3,5-di-tert-butylcatechol (3,5-dtbc) and can thus be considered as a promiscuous man-made catalyst.
Experimental
Materials and measurements
All reagents of high purity grade were purchased from commercial sources and used as received. In the characterization of H2bpbpmp and its complexes, and in the kinetic studies, spectroscopic grade solvents from Merck, dried using molecular sieves, were used.
The potentiometric studies were carried out with a Micronal 375 pH meter fitted with blue-glass and calomel reference electrodes calibrated to read log[H+] directly, designated as pH. Bidistilled water in the presence of KMnO4 was used to prepare CH3CN (1:1 v/v) solutions. The electrode was calibrated using the data obtained from a potentiometric titration of a known volume of a standard 0.0100 mol L-1 HCl solution with standard 0.100 mol L-1 KOH. The temperature was kept at 25.00 ± 0.05 ºC, and the ionic strength was adjusted to 0.100 mol L-1 by addition of KCl. The experimental solutions were titrated with 0.100 mol L-1 standard CO2-free KOH. Equilibrium measurements were taken using solutions containing 0.05 mmol of complex. The experiments were performed on 50.00 mL of the CH3CN:H2O (1:1 v/v) solution in a thermostated cell purged with argon, using 0.100 mol L-1 KOH solutions. The experiments were carried out in triplicate. Computations of the triplicates were all carried out with the BEST7 program, and the species diagrams were obtained with SPE and SPEPLOT programs.36
Complexes 1 and 2 were also titrated spectrophotometrically. All titrations were carried out under an argon atmosphere at 25 ºC in 1:1 (v/v) CH3CN:H2O. The pH was first adjusted with HCl and the complex titrated with successive additions of CO2 free KOH 0.124 mol L-1. At each measured pH, an aliquot was removed to record the UV-Vis spectrum.
A dark red crystal was selected from the sample of complex 1 and fixed at the end of a glass fiber for X-ray analysis. Crystallographic measurements of complex 1 were carried out with an Enraf-Nonius CAD4 diffractometer with graphite-monochromated Mo-Kα radiation, at room temperature. Cell parameters were determined from 25 carefully centered reflections using a standard procedure.37 All data were corrected for Lorentz and polarization effects.38 A semi-empirical absorption correction based on the azimuthal scans of seven appropriate reflections was also applied to the collected intensities with the PLATON program,39,40 with maximum and minimum transmission factors of 0.873 and 0.791, respectively. The structure was solved by direct methods and refined by full-matrix least-squares methods using SIR9741 and SHELXL9742 programs, respectively. All non-hydrogen atoms were refined anisotropically. H atoms attached to C atoms were placed at their idealized positions, with C-H distances and Ueq values taken from the default settings of the refinement program. The water molecule H atoms were found from a Fourier difference map and treated with the riding model. Two oxygen atoms of the perchlorate counterion are disordered over two alternative positions. The site occupancies of 0.555(14) and 0.445(14) for the disordered atoms were refined and the geometric parameters around the chlorine atom were constrained. Further selected crystallographic data are summarized in Table 1.
Syntheses
The unsymmetric dinucleating ligand H2bpbpmp was prepared accordingly to procedures previously described in the literature.23
[(bpbpmp)FeIII(m-OAc)2FeII](ClO4)H2O, (1)
0.5 mmol of H2bpbpmp were dissolved in 10 mL of CH3CN under argon. To this solution were added, respectively, 1 mmol of Fe(ClO4)26H2O and 2 mmol of NaOAc in MeOH (10 mL) also under argon. The resulting solution became deep purple. Finally, 5 mL of diisopropyl ether in the presence of air were added to the purple solution, which was kept in the refrigerator. After 24 h crystals suitable for X-ray crystallography were formed, which were collected by filtration (0.32 g, 70 %). Elemental Analysis for C38H41ClFe2N5O11 (890.91 g mol-1): Found C, 51.56; H, 4.60; N, 7.78. Calculated C, 51.23; H, 4.64; N, 7.86%. The compound is stable in air in the solid state. (Caution! As with all perchlorate salts, samples should be handled with care.)
[(bpbpmp)FeIII(m-OAc)2FeIII](ClO4)23CH3OH3H2O (2)
To a solution of Fe(ClO4)39H2O (2.0 mmol) in 20 mL of methanol, sodium acetate trihydrate (2.5 mmol) and H2bpbpmp (1.07 mmol) were added. The clear dark blue solution was heated to 40 ºC and stirred for 10 min at ambient atmosphere. After cooling of the solution to room temperature, a blue microcrystalline precipitate was formed, which was filtered off and washed with 2-propanol and ether (0.73 g, 65%). Elemental Analysis for C41H57Cl2Fe2N5O20 (1121.17 g mol-1): Found C, 44.3; H, 4.58; N, 6.24. Calculated C, 43.87; H, 5.12; N, 6.24%.
Kinetics
Catecholase-like activity toward the oxidation of the model substrate 3,5-di-tert-butylcatechol (3,5-dtbc) was investigated. The experiments were followed spectrophotometrically on a Varian Cary50 Bio spectrophotometer connected to a computer, by monitoring the increase in the 3,5-di-tert-butyl-quinone (3,5-dtbq) characteristic absorption band at 400 nm over time. The conversion of the reaction rate units was carried out using ε = 1760 L mol-1 cm-1 (measured in the reaction conditions) for 3,5-di-tert-butylquinone, and the initial concentration of complex.
The experiments were carried as follows: a stock solution of the complex was prepared in methanol (16 h earlier), saturated with argon (4.4×10-4 mol L-1). A stock solution of the substrate was also prepared in methanol ([3.5-dtbc] = 8.26×10-2 mol L-1). To a 4 mL cuvette, 100 µL of argon-saturated buffer and 100 µL of 1 were added. Subsequently, O2-saturated methanol, deionized water and substrate (20-200 µL) were added to complete 3.0 mL (giving the proportion 1.8 mL methanol : 1.2 mL water, v/v).
The initial rate was obtained from the slope of the absorbance vs. time plot over the first 10 min of reaction. Each experiment was performed at least three times, and the obtained plots were analyzed by the initial rate method. A kinetic treatment using nonlinear Michaelis-Menten approach was applied.
The influence of the pH on the catalyzed oxidation of the 3,5-di-tert-butylcatechol was determined over the pH range of 4.4 to 9.0. Reactions were performed using the following conditions in the cuvette: air saturated CH3OH/H2O, 3:2 v/v, [1] = 1.39×10-5mol L-1, [3,5-dtbc] = 2.77×10-3 mol L-1, [buffer] = 3.3×10-2 mol L-1 (Buffer = MES, 2-[N-morpholine]ethanesulfonic acid, or TRIS, tris(hydroxymethyl)aminomethane). Dependence of the reaction rates on [3,5-dtbc] was determined at pH 6.0, in a methanol:water (3:2, v/v) solution. Conditions: [1] = 1.46×10-5 mol L-1; [3,5-dtbc] = 0.5-5.5×10-3 mol L-1, [buffer] = 3.3×10-2 mol L-1 (buffer = MES).
To determine which reduced oxygen species (hydrogen peroxide or water) was formed during the reaction, mixtures were prepared as in the kinetics experiments. After 1 h of reaction an equal volume of H2SO4 (5×10-3 mol L-1) was added. The quinone was washed twice with dichloromethane (2×3 mL). To a 2 mL aliquot of the aqueous layer a KI solution (1 mL, 0.30 mol L-1) was added. Finally, ammonium molybdate (0.1 mol L-1) solution was added in catalytic amounts to specifically accelerate the formation of I3 in the presence of H2O2. The increase in the I3- absorption band at 353 nm (ε = 26,000 L mol-1 cm-1) was monitored by UV-Vis spectroscopy.43-45 The reaction without oxygen was prepared under the kinetic conditions and the absorbance increase at 400 nm due to quinone release was not observed.
DNA extraction
Plasmid DNA (pBSKII) (Stratagene) was produced in Escherichia coli and purified with HiSpeed Maxiprep Kit (Qiagen) following the protocol described by the manufacturer.
DNA cleavage under anaerobic and aerobic conditions
Complex 1 solutions were diluted in acetonitrile in an argon-filled glove bag to avoid Fe oxidation. A two-step procedure was used to obtain deoxygenated water, piperazine-1,4-bis(2-ethanesulfonic acid) (PIPES) buffer and ethylenediaminetetraacetic acid (edta). A parallel experiment with the oxidative cleavage system [Fe(edta)]2/DTT (dithiothreitol) was used to demonstrate the deoxygenation procedure efficiency.46,47 DNA cleavage experiments were performed in a final volume of 20 µL containing 35 µmol L-1 bp (DNA base pairs) pBSK II plasmid DNA, complex 1 [40, 80 and 160 µmol L-1], 25% acetonitrile (v/v) and 25 mmol L-1 PIPES buffer, pH 6.0. Reaction mixtures were incubated at 50 ºC for 2 h in a sealed argon-filled vacuum desiccator.
Identical complex 1 solutions as prepared in argon-filled bags were used for the reactions under aerobic conditions. All other conditions and procedures were essentially the same as those described for anaerobic reactions.
Reactions were stopped by chilling tubes on ice and adding 4 µL of ice-cold loading-buffer (50% glycerol, 0.2 mol L-1 edta, 0.05% bromophenol blue). Samples were submitted to agarose gel electrophoresis and stained with ethidium bromide. Resulting gels were digitalized with a photo-documentation system (UVP, CA, USA) and DNA bands were quantified using LabWorks software version 4.0 (UVP).
Kinetics DNA assays
DNA cleavage reactions were performed at 50 ºC in 25 mmol L-1 PIPES (pH 6.0), using 35 µmol L-1 bp (DNA base pairs) pBSK II plasmid DNA at concentrations of complex 1 ranging from 0 to 160 µmol L-1, in a final volume of 160 µL. Aliquots of 20 µL were withdrawn at different time intervals (0-60 min) and were analyzed as described for the DNA cleavage.22, 48
Reaction mechanism
Cleavage experiments to understand the reaction mechanism were carried out with potential inhibiting agents such as a hydroxyl radical scavenger dimethyl sulfoxide (dmso), a singlet oxygen scavenger (sodium azide, NaN3) and a minor groove binder (distamycin).7,49-51
Reactions in the presence of the above compounds were carried out in 20 µL final volume, containing: 35 µmol L-1 bp (plasmid DNA), in 25 mmol L-1 PIPES, pH 6.0, for 1 h at 50 ºC (other conditions are described in the Figure legends). Reactions were stopped by chilling tubes on ice and adding 4 µL of ice-cold loading-buffer. Samples were analyzed in 0.8% agarose gel.
Results and Discussion
Synthesis of 1 and 2 and crystal structure of 1
Reaction of one equivalent of the unsymmetrical ligand H2bpbpmp with two equivalents of [Fe(OH2)6](ClO4)2 in the presence of NaOAc in a 1:1 mixture of CH3CN and methanol results in the formation of the title complex [(bpbpmp)FeIII(µ-OAc)2FeII](ClO4)H2O (1). The properties determined by elemental, UV-Vis and electrochemical analysis are in agreement with the composition previously proposed for 1.29 Crystals of 1 suitable for X-ray crystallography were obtained by slow diffusion of diisopropyl ether in a methanolic solution of the title complex. On the other hand, when the reaction of one equivalent of H2bpbpmp was carried out with two equivalents of iron(III) perchlorate in the presence of excess perchlorate, the oxidized dinuclear complex [(bpbpmp)FeIII(µ-OAc)2FeIII](ClO4)3CH3COH3H2O (2) was obtained. This complex was characterized by elemental analysis, electronic spectroscopy, and electrochemistry/spectroelectrochemistry. All attempts to obtain suitable crystals of 2 for X-ray analysis were unsuccessful.
The structure of complex 1 consists of discrete dinuclear [FeIII(bpbpmp)(µ-OAc)2FeII]+ cations, uncoordinated perchlorate anions and one molecule of water in the crystallographic asymmetric unit. An ORTEP drawing of 1 with its atom-labeling scheme is shown in Figure 1. Selected bond distances and angles are given in Table 2.
The structure of the cation [FeIII(bpbpmp)(µ-OAc)2FeII]+ in 1 is similar to that determined for the corresponding BF4- salt.31 Briefly, in the structure of [FeIII(bpbpmp)(µ-OAc)2FeII]+ the metal centers are bridged by the phenolate oxygen atom of the bpbpmp2- ligand and by two exogenous acetate ligands, as has been observed in numerous isostructural complexes containing the same ligand.22-29 As expected for a dinuclear mixed-valence complex containing the unsymmetrical bpbpmp2- ligand, the coordination sphere of the FeIII is completed by two nitrogen atoms from the tertiary amine (N1) and pyridyl (N22) groups, and by a terminal phenolate oxygen (O10), while the FeII is on the softer side of bpbpmp2-, coordinated by three nitrogen atoms, pyridyl N32 and N42 and tertiary amine N2.
The bridging Ophenolate is coordinated unsymmetrically to the two metal centers, with the bond distances FeIII-O = 2.005(3) and FeII-O = 2.102(3) Å being slightly longer than those observed in the mixed-valence complex [FeIIIFeII(bpmp)(OPr)2]2+ (FeIII-O = 1.943(2) and FeII-O1 = 2.090(2) Å) containing the symmetric Hbpmp (2,6-bis[(bis(2-pyridylmethyl)amino)methyl]-4-methylphenol) ligand.29 As in the analogous FeIIIMII complexes (MII = Zn, Cu, Ni, Mn), the terminal phenolate coordinated to the FeIII is trans to the bridging phenolate. The metal-metal distance (3.5041(13) Å) as well as the FeIII-O-FeII angle (117.14(13)º) in the FeIIIFeII complex are similar to those observed in all of the isostructural FeIIIMII complexes (3.470-3.510 Å and 115-119º).22-29 Additionally, the terminal phenoxoFeIII-Ophenolate bond length (1.902(3) Å) in 1 is slightly longer than the corresponding bond in the FeIIIZnII complex (1.890 Å).52
Solution studies of 1 and 2
Potentiometric titration studies of the complex [FeIII(bpbpmp)(µ-OAc)2FeII]ClO4(1) in 1:1 acetonitrile/buffer have been described previously and showed the presence of four protonation equilibria, two of which (pKa2 = 4.13 and pKa4 = 7.53) are catalytically relevant for the hydrolysis of phosphate diester bonds.31 Results are summarized in Table 3, with the kinetic pKas of 1 for comparison.
Considering that the oxidized form of complex 2 in aqueous solution is the origin of the catalytically active species for the oxidation of catechols (vide infra), we also carried out potentiometric titration of this complex under experimental conditions identical to those employed for complex 1. As can be observed in Figure 2, four mol of KOH were neutralized in the pH range of 2.4-10.9. Fitting the data with the BEST program results in the following deprotonation constants: pKa2 = 3.14, pKa3 = 4.22, pKa4 = 6.45 and pKa5 = 9.65. It is well known that in related MIII(µ-OAc)2MII dinuclear complexes, the bridging carboxylate groups dissociate resulting in the formation of FeIII-OH/OH2 species when in aqueous solution. Thus, the four-deprotonation/protonation equilibrium steps for complex 2 are proposed in Scheme 2 with the corresponding species distribution given in Figure 2. It is assumed that when 2 is dissolved in aqueous solution at pH 2.4, two acetate groups are released, leading to the [(OH2)FeIII(µ-OH) FeIII(OH2)] species (structure A in Scheme 2).
Deprotonation of the water molecule (pKa2 = 3.14) coordinated to the FeIII center containing the softer terminal tridentate group (one amine and two pyridine) leads to the formation of the [(OH2)FeIII(µ-OH)FeIII(OH)] species (structure B in Scheme 2). This observation is based mainly on the fact that the FeIII center bound by three nitrogens of bpbpmp2- is a stronger Lewis acid than the FeIII center coordinated by the harder N2O-donor pendant arm containing the terminal phenolate group. The pKa3 of 4.22 is attributed to dissociation of the bridging µ-OH group which results in the formation of [(OH2)FeIII(µ-O)FeIII(OH)] (structure C in Scheme 2) as one of the catalytically active species in the oxidation of catechols (vide infra). This pKa value is around 1.5 pH units lower than the corresponding pKa determined in the mixed-valence FeIIIFeII complex 1 and can be explained in terms of a higher Lewis acidity exerted by two FeIII centers on the bridging µ-OH group in complex 2. Deprotonation of the water molecule (pKa4 = 6.45) terminally coordinated to the FeIII center which contains the harder tridentate N2O-donor group (one amine, one pyridine and one phenolate) leads to the formation of the [(OH)FeIII(µ-O)FeIII(OH)] species (structure D in Scheme 2) which is the most active catalyst in the oxidation of catechols. Again, as expected, this pKa4 = 6.45 is approximately one pH unit lower when compared to the protonation/deprotonation equilibrium found for the corresponding FeII-OH2 bond in the mixed-valence complex 1. Moreover, it is also important to note that this pKa4 value is in good agreement with the pKa of 7.0 determined for the dissociation of the second FeIII-bound water molecule in a dinuclear FeIII complex which contains a similar NO-donor set as that proposed for the [(OH2)FeIII(µ-O)FeIII(OH)] species.53 Finally, a fourth deprotonation/protonation equilibrium at pKa5 = 9.65 was observed, despite the fact that in the [(OH)FeIII(µ-O)FeIII(OH)] species there appears to be no ionizable protons under these experimental conditions. We tentatively suggest the substitution of one pyridine group bound to one of the FeIII centers by one water molecule with subsequent deprotonation of this water molecule as being responsible for this equilibrium.53
UV-visible spectroscopy is another tool employed to identify species in equilibrium and can be very useful to assign the catalytically active species in solution. Moreover, it should be noted that complexes 1 and 2 display ligand-to-metal charge transfer (LMCT) processes which are very sensitive to any electronic change (e.g. Lewis acidity) in the metal centers. Thus, we used electronic spectroscopy to further confirm the catalytically active species of 1 and 2 under kinetic conditions.
Spectrophometric titration of the mixed-valence complex 1 reveals four protonation/deprotonation constants which are comparable to those obtained from the potentiometric experiments (Table 3, Figure S1).31 In general, hypsochromic shifts are detected (Figure 3) when the pH of the solution rises, suggesting that the replacement of FeIII-OH2 by FeIII-OH, or of FeIII(µ-OH)FeIII by FeIII(µ-O)FeIII in the systems in equilibrium does affect the energy of the LMCT processes. This is probably due to a lower order of Lewis acidity of the FeIII-centers when the stronger donor hydroxo or oxo groups are coordinated, so that the t2g orbitals in these species are at higher energy than the t2g orbitals in the FeIII-OH2 complexes. Similarly, spectrophotometric titration of the oxidized form of complex 2 also demonstrates that the λmax of the LMCT is strongly influenced by the pH of the solution (Figure S2, Figure S3). However, in this case only three reliable pKa values (Table 3) could be obtained in the pH range of 2.4-10.9, which are in good agreement with those calculated in the potentiometry studies.
Kinetics
In recent years, it has been demonstrated that many enzymes are able to catalyze distinctly more than one chemical reaction. This ability, called catalytic promiscuity, may be related to different kinds of bond formation or cleavage or different catalytic pathways or even both.1,54 Biomimetic models can be more susceptible to catalytic promiscuity, since they do not have a complex structure as do enzymes. In this regard, biomimetic models are expected to be much more accessible to different substrates and more dependent on the environmental conditions.
The catalytic promiscuity of the complex [FeIII(bpbpmp)(m-OAc)2FeII]ClO4, (1), was evaluated by testing its hydrolase-like activity in the cleavage of DNA and its catecholase-like activity toward the oxidation of the model substrate 3,5-di-tert-butylcatechol (3,5-dtbc).31 The phosphatase-like activity of 1 in the hydrolysis of the model substrate bis-2,4-dinitrophenylphosphate (2,4-bdnpp) has been demonstrated very recently.31
The catecholase activity of complexes 1 and 2 was determined using the substrate 3,5-di-tert-butylcatechol by following spectrophotometrically the absorbance increase due to the formation of 3,5-di-tert-butylquinone (λmax = 400 nm), under conditions of excess substrate. It is important to note that there is no reaction (not even one cycle) using 1 as the catalyst during a period of three hours in the absence of oxygen. However, after this time, if the cuvettes were opened to allow the entrance of oxygen, the reaction begins immediately. Therefore, the kinetics data obtained suggest the formation of an oxidized form of FeIIIFeIII as the catalyst, which can be obtained through oxidation of 1 in the presence of air or through chemical oxidation of 1 with H2O2 or from the dissolution of the complex 2 in kinetic conditions. Indeed, the spectrum of 1 under kinetic conditions without the substrate shows the disappearance of the intervalence band centered at 1100 nm, indicating that the species [(OH2)FeIII(µ-O)FeII(OH)] is oxidized to complex[(OH2)FeIII(µ-O)FeIII(OH)]in the presence of air (Figure S4).
The pH-dependence of the oxidation reaction of 3,5-dtbc was evaluated from pH 4.4 to 9.0 and, as can be observed in Figure 4, there are two distinct kinetic behaviors indicating the presence of different catalysts. Initially, the reaction rate increases only slightly and reaches a plateau in the pH range 6.0-6.5. A further increase in pH results in an almost uninterrupted increase in the initial rate V0, suggesting that a second species is acting as the catalyst. However, from the inflection points in the plot given in Figure 4, the pKas at 5.0 and 6.7 are only tentatively attributed given the small increase in reactivity in the pH range 4.3-6.5 and the poor definition of the plateau at pH 7.0. Even so, these pKa values are in relatively good agreement with those obtained from the potentiometric and spectrophotometric experiments (4.2 and 6.5), thus suggesting that the [(OH2)FeIII(µ-O)FeIII(OH)] (C) and [(OH)FeIII(µ-O)FeIII(OH)] (D) in Scheme 2 are most probably the catalytically active species in the oxidation of 3,5-dtbc. Indeed, the observed pH dependence of the reaction may be due to the successive deprotonation of FeIII-coordinated water molecules, and probably the FeIII-OH groups are able to interact with catechol promoting the deprotonation of the substrate and concomitant coordination to the metal centers.
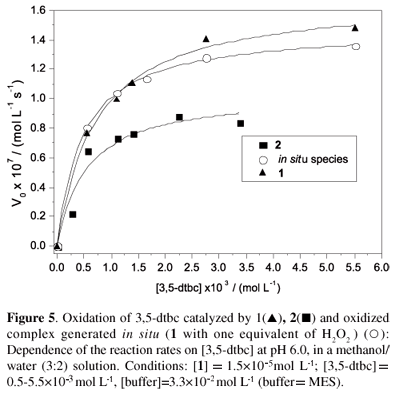
The dependence of the initial rates on the concentration of 3,5-dtbc using complex 1 as the catalyst was investigated at 25 ºC at pH 6.0 and revealed saturation kinetics with Michaelis-Menten-like behavior (Figure 5).
The kinetic parameters (Vmax=1.69×10-7 mol L-1 s-1, KM=7.1×10-4 mol L-1 and kcat=11.6×10-3 s-1), where KM and kcat are the Michaelis-Menten and the catalytic constants, respectively, were obtained from Michaelis-Menten nonlinear treatment (V0vs. [3,5-dtbc]). The kinetic parameters obtained for the reaction catalyzed by 2 are in the same order as those found for complex 1 in the presence of oxygen or H2O2 (Table 4), thus supporting the mechanistic proposal that the FeIIIFeIII species must be present to initiate the catalytic cycle. Moreover, accumulation of H2O2 during turnover was confirmed by means of the molybdate-accelerated I3- assay, which indicates that reoxidation of the FeIIFeII species back to the active FeIIIFeIII species occurs with a 1:1 (O2:3,5-dtbc) stoichiometry and concomitant formation of hydrogen peroxide.
Finally, papers found in the literature related to catechol artificial biomimetics generally refer to binuclear CuII complexes.43-45,55 It is interesting to note that the kinetic parameters obtained in this study for complex 1 orcomplex 2 are of the same order as those found for CuIICuII complexes with similar ligands, but with 1 or 2 being active at lower pH values (6.0 vs. 8-9),43-45 which is a consequence of the higher Lewis acidity of FeIII when compared to CuII.
Considering all of the experimental data obtained, the mechanism shown in Scheme 3 is proposed.
DNA cleavage
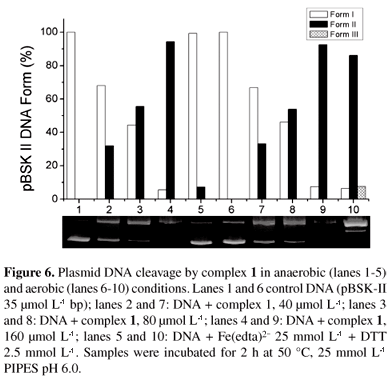
To verify the ability of complex 1 to cleave DNA phosphodiester bonds, we tested its activity in the absence and in the presence of oxygen. Data from the experiments under aerobic and anaerobic conditions (Figure 6) suggest that a hydrolytic mechanism is responsible for the DNA cleavage, since the reaction is not affected by the absence of oxygen. The control experiment with Fe(edta)2/dithiothreitol demonstrated that there is no residual oxygen because DNA cleavage was totally inhibited under anaerobic conditions.
It is also clear from Figure 6 that 1 was able to cleave plasmid DNA in a concentration-dependent manner. An increase in circular (nicked, form II) plasmid DNA, and a proportional decrease in supercoiled DNA (form I) was observed with increasing complex concentration.
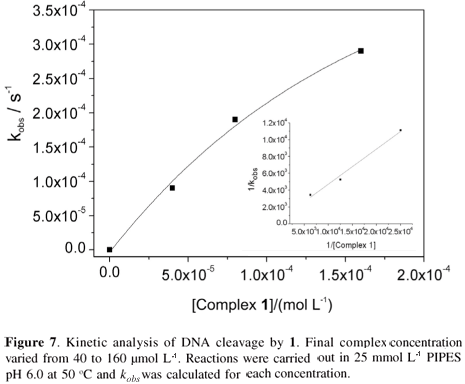
Pseudo-Michaelis-Menten conditions were used in the kinetic studies of the DNA cleavage by 1.48,56,57 We observed that the decrease in supercoiled DNA fitted a single exponential decay curve (Figure 7). Cleavage rates at various concentrations of 1 (40-160 µmol L-1) and a constant DNA concentration were calculated and a plot of kobs versus the concentration of 1 shows saturation behavior. Lineweaver-Burk linearization of these data (inset Figure 7) gives the following kinetics parameters: kcat 1.91×10-3 s-1; KM 7.96×10-4 mol L-1; kcat/KM 2.40 mol s-1 L-1, providing a rate enhancement of 1.91×108 over the uncatalyzed double-strand DNA cleavage.
Figure 8 presents the results of DNA cleavage by 1 in the presence of potential inhibiting agents: a hydroxyl radical scavenger (dmso), a singlet oxygen scavenger (sodium azide, NaN3) and a DNA minor groove binder (distamycin). Neither dmso nor sodium azide interfered with the cleavage activity of the complex. These results are in agreement with those found under anaerobic conditions, demonstrating that 1 cleaves DNA hydrolytically and that oxygen radicals are not involved in the reaction. Also in Figure 8, the activity of 1 is not inhibited by distamycin, suggesting that 1 interacts with double strand DNA through the major grove.
Conclusions
The complexes [(bpbpmp)FeIII(µ-OAc)2FeII](ClO4) (1) and [(bpbpmp)FeIII(µ-OAc)2FeIII](ClO4)2 (2) containing the unsymmetrical dinucleating ligand H2bpbpmp, 2-{[(2-hydroxybenzyl)(2-pyridylmethyl)aminomethyl]-4-methyl-6-[bis(2-pyridylmethyl)aminomethyl]}phenol, were synthesized and their properties and catalytic features explored. The X-ray structure of 1 reveals an {FeIII(µ-Ophenolate)(µ-OAc)2FeII} core in which the FeIII and FeII metal centers are, respectively, bound by the hard and soft side of the bpbpmp2- ligand. Kinetic studies on the cleavage of double strand DNA (dsDNA) by 1 at the optimum pH (6.0) and 50 ºC show Michaelis-Menten behavior with a rate enhancement of 1.9×108 over the uncatalyzed reaction. Data from the experiments under aerobic and anaerobic conditions along with kinetic studies on a model diester substrate suggest a hydrolytic mechanism in which a terminal FeIII-bound hydroxide is the initiating nucleophile in the catalytic reaction. On the other hand, when the [(bpbpmp)(HO)FeIII(µ-OH)FeII(H2O)]+ species is exposed to air, the oxidized FeIIIFeIII complex is formed. This complex shows significant catecholase activity in the oxidation of 3,5-di-tert-butylcatechol (methanol/water buffer, 60:40, pH = 5.8) and the reaction follows Michaelis-Menten behavior with kcat = 1.16×10-2 s-1 and KM = 7.1×10-4 mol L-1. The complex is thus revealed to be an important example of a synthesized promiscuous catalyst, with activity in both the oxidation of catechols and the hydrolysis of phosphodiester bonds.
Supplementary Information
Figures S1-S4 are available free of charge at http://jbcs.sbq.org.br, as a PDF file. Crystallographic data (excluding structural factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 747619. Copies of the data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK ; fax: +44 1223 336033 ; or e-mail: deposit@ccdc.cam.ac.uk).
Acknowledgments
The authors thank CNPq, CAPES, FAPESC and INCT-Catálise for financial support. G. Schenk and L. Gahan acknowledge funds awarded by the Australian Research Council through grants DP0986292, DP0986613, DP0664039 and DP0558652.
References
1. Kazlauskas, R. J.; Curr. Opin. Chem. Biol. 2005, 9, 195.
2. Bornscheuer, W. T.; Kazlauskas, R. J.; Angew. Chem. 2004, 43, 6032.
3. O'Brian, P. J.; Herschlag, D.; Chem. Biol. 1999, 6, 91.
4. Taglieber, A.; Höbenreich, H.; Carballeira, J. D.; Mondière, R. J. G.; Reetz, M. T.; Angew. Chem. 2007, 46, 8597 and references therein.
5. O'Brian, P. J.; Chem. Rev. 2006, 106, 720 and references therein.
6. Olguin, L. F.; Askew, S. E.; O'Donoghue, A. C.; Hollfelder, F.; J. Am. Chem. Soc. 2008, 130, 16547.
7. Zalatan, J. G.; Herschlag, D.; J. Am. Chem. Soc. 2006, 128, 1293.
8. Hemrika, W.; Renirie, R.; Dekker, H. L.; Barnett, P.; Wever, R.; Proc. Natl. Acad. Sci. USA 1997, 94, 2145.
9. Yoneyama, T.; Shiozawa, M.; Nakamura, M.; Suzuki, T.; Sagane, Y. Katoh, Y.; Watanabe, T.; Ohyama, T.; J. Biol. Chem. 2004, 279, 37477.
10. Bozzo, G. G.; Raghothama, K. G.;. Plaxton, W. C.; Eur. J. Biochem. 2002, 269, 6278.
11. Kenton, P.; Mur, L. A. J.; Draper, J.; J. Exp. Bot. 1999, 50, 1331.
12. Jakobek, J. L.; Lindgren, P. B.; J. Exp. Bot. 2002, 53, 387.
13. Oliveira, M. C. B.; Scarpellini, M.; Neves, A.; Terenzi, H.; Bortoluzzi, A. J.; Szpoganicz, B.; Greatti, A.; Mangrich, A. S.; Souza, E. M.; Fernandez, P. M.; Soares, M.; Inorg. Chem. 2005, 44, 921.
14. Rey, N. A.; Neves, A.; Bortoluzzi, A. J.; Pich, C. T.; Terenzi, H.; Inorg. Chem. 2007, 46, 348.
15. Deck, K. M.; Tseng, T. A.; Burstyn, J. N.; Inorg. Chem. 2002, 41, 669.
16. Hegg, E. L.; Burstyn, J. N.; J. Am. Chem. Soc. 1995, 117, 7015.
17. Hegg, E. L.; Burstyn, J. N.; Inorg. Chem. 1996, 35, 7474.
18. Parrilha, G. L.; Fernandes, C.; Bortoluzzi, A. J.; Szpoganicz, B.; Silva, M. S.; Pich, C. T.; Terenzi, H.; Horn, A., Jr.; Inorg. Chem. Commun. 2008, 11, 643.
19. Mitic, N.; Smith, S. J.; Neves, A.; Guddat, L. W.; Gahan, L. R.; Schenk, G.; Chem. Rev. 2006, 106, 3338.
20. Gavrilova, A. L.; Bosnich, B.; Chem. Rev. 2004, 104, 349;
21. Gahan, L. R.; Smith, S. J.; Neves, A.; Schenk, G.; Eur. J. Inorg. Chem. 2009, 2745.
22. Neves, A.; Lanznaster, M.; Bortoluzzi, A. J.; Peralta, R. A.; Casellato, A.; Castellano, E. E.; Herrald, P.; Riley, M. J.; Schenk, G.; J. Am. Chem. Soc. 2007, 129, 7486.
23. Karsten, P.; Neves, A.; Bortoluzzi, A. J.; Drago, V.; Lanznaster, M.; Inorg. Chem. 2002, 41, 4624.
24. Karsten, P.; Neves, A.; Bortoluzzi, A. J.; Strähle, J.; Maichle-Mössmer, C.; Inorg. Chem. Commun. 2002, 5, 434.
25. Batista, S. C.; Neves, A.; Bortoluzzi, A. J.; Vencato, I.; Peralta, R. A.; Szpoganicz, B.; Aires, V. E.; Severino, P. A.; Inorg. Chem. Commun. 2003, 6, 1161.
26. Lanznaster, M.; Neves, A.; Bortoluzzi, A. J.; Aires, V. V. E.; Szpoganicz, B.; Terenzi, H.; Severino, R. C.; Fuller, J. M.; Drew, S. C.; Gahan, L. R.; Hanson, G. R.; Riley, M. J.; Schenk, G.; J. Biol. Inorg. Chem. 2005, 10, 319.
27. Schenk, G.; Peralta, R. A.; Batista, S. C.; Bortoluzzi, A. J.; Szpoganicz, B.; Dick, A. K; Herrald, P.; Hanson, G. R.; Szilagyi, R. K.; Riley, M. J.; Gahan, L. R.; Neves, A.; J. Biol. Inorg. Chem. 2008, 13, 139.
28. Smith, S. J.; Casellato, A.; Hadler, K. S.; Mitić, N.; Riley, M. J.; Bortoluzzi, A. J.; Szpoganicz, B.; Schenck, G.; Neves, A.; Gahan, L.; J. Biol. Inorg. Chem. 2007, 12, 1207.
29. Neves, A.; Brito, M. A.; Drago, V.; Griesar, K.; Haase, W.; Inorg. Chim. Acta 1995, 237, 131.
30. Gonçalves, N. S.; Horn Jr., A.; Lanznaster, M.; Noda, L. K.; Neves, A.; J. Braz. Chem. Soc. 2006, 17, 1658.
31. Smith, S. J.; Jovito, R.; Horn Jr., A.; Nascimento, O. R.; Noble, C. J.; Hanson, G. H.; Stranger, R.; Jayaratne, V.; Cavigliasso, G.; Gahan, L. R.; Schenk, G.; Neves, A.; Riley, M. J.; unpublished results.
32. de Oliveira, I. R.W. Z.; Neves, A.; Vieira, I. C.; Sens. Actuators 2008, B 129, 424.
33. Bernhardt, P. V.; Schenk, G.; Wilson, G.; J. Biochem. 2004, 43, 10387.
34. Wang, D. L.; Holz, R. C.; David, S. S.; Que Jr., L.; Stankovich, M. T.; Biochemistry 1991, 30, 8187.
35. Bunton, C. A.; Farber, S. J.; J. Org. Chem. 1969, 34, 767.
36. Martell, A. E.; Motekaitis, R. J.; Determination of Stability Constants, 2nd ed., VHC Publishers: Weinheim, Germany, 1992.
37. Enraf-Nonius; CAD-4 EXPRESS; Version 5.1/1.2; Enraf-Nonius: Delft, The Netherlands, 1994.
38. Spek, A.L.; HELENA; CAD-4 Data Reduction Program; Univ. of Utrecht, The Netherlands, 1996.
39. Spek, A.L.; Acta Crystallogr. 1990, A46, C34.
40. North, A. C. T.; Phillips, D. C.; Mathews, F. S.; Acta Crystallogr. 1968, A24, 351.
41. Altomare, A.; Burla, M. C.; Camalli, M.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R.; J. Appl. Cryst. 1999, 32, 115.
42. Sheldrick, G.M.; Acta Crystallogr. 2008, A64, 112.
43. Neves, A.; Rossi, L. M.; Bortoluzzi, A. J.; Szpoganicz, B.; Wiezbicki, C.; Schwingel Haase, E.; Ostrovsky, W. S.; Inorg. Chem. 2002, 41, 1788.
44. Peralta, R. A.; Neves, A.; Bortoluzzi, A. J.; dos Anjos, A.; Xavier, F. R.; Szpoganicz, B.; Terenzi, H.; de Oliveira, M. C. B.; Castellano, E.; Friedermann, G. R.; Mangrich, A. S.; Novak, M. A.; J. Inorg Biochem. 2006, 100, 992.
45. Neves, A.; Rossi, L. M.; Bortoluzzi, A. J.; Haase, W.; Werner. R.; J. Braz. Chem. Soc. 2001, 12, 747.
46. Gonzalez-Alvarez, M.; Alzuet, G.; Borras, J.; Macías, B.; Del Olmo, M.; Liu-Gonzalez, M. E.; Sanz, F.; J. Inorg. Biochem. 2002, 89, 29.
47. Scarpellini, M.; Neves, A.; Hörner, R.; Bortoluzzi, A. J.; Szpoganicz, B.; Zucco, C.; Nome, R. A.; Drago, V.; Mangrich. A.S.; Ortiz, W. A.; Passos, W.A.; de Oliveira, M. C.; Terenzi, H.; Inorg. Chem. 2003, 42, 8353.
48. Sreedhara, A.; Freed, J. D. E.; Cowan, J. A.; J. Am. Chem. Soc. 2000, 122, 8814.
49. Brezova, V.; Valko, M.; Breza, H.; Morris, H.; Telser, J.; Dvoranova, D.; Kaiserova, K.; Varecka, L.; Mazur, M.; Leibfritz, D.; J. Phys. Chem. B 2003, 107, 2415.
50. Macías, B.; Villa, M.V.; Gómez, B.; Borrás, J.; Alzuet, G.; González-Álvarez, M.; Castineiras, A.; J. Inorg. Biochem. 2007, 101, 444.
51. Uytterhoeven, K.; Sponer, J.; Van Meervelt, L.; Eur. J. Biochem. 2002, 269, 2868.
52. Lanznaster, M.; Neves, A.; Bortoluzzi, A. J.; Szpoganicz, B.; Schwingel, E.; Inorg. Chem. 2002, 41, 5641.
53. Horn Jr, A.; Vencato, I.; Bortoluzzi, A.J.; Hörner, R.; Silva, R.A.N.; Szpoganicz, B.; Drago, V.; Terenzi, H.; Oliveira, M.C.B.; Werner, R.; Haase. W.; Neves, A.; Inorg. Chim. Acta 2005, 358, 339.
54. Bornscheuer, U. T.; Kazlauskas, R. J.; Angew. Chem. Int. Ed. 2004, 43, 6032.
55. Smith, S. J.; Noble, C. J.; Palmer, R. C.; Hanson, G. R.; Schenk, G.; Gahan, L. R.; Riley, M. J.; J. Biol. Inorg. Chem. 2008, 13, 499.
56. Cowan, J. A.; Curr. Opin. Chem. Biol. 2001, 5, 634.
57. Sreedhara, A.; Cowan, J. A.; J. Biol. Inorg. Chem. 2001, 6, 337.
Received: October 1, 2009
Web Release Date: March 15, 2010
This paper is dedicated to our friend Professor Icaro Moreira in memoriam
Supplementary Information
- 1. Kazlauskas, R. J.; Curr. Opin. Chem. Biol. 2005, 9, 195.
- 2. Bornscheuer, W. T.; Kazlauskas, R. J.; Angew. Chem. 2004, 43, 6032.
- 3. O'Brian, P. J.; Herschlag, D.; Chem. Biol. 1999, 6, 91.
- 4. Taglieber, A.; Höbenreich, H.; Carballeira, J. D.; Mondière, R. J. G.; Reetz, M. T.; Angew. Chem. 2007, 46, 8597 and references therein.
- 5. O'Brian, P. J.; Chem. Rev 2006, 106, 720 and references therein.
- 6. Olguin, L. F.; Askew, S. E.; O'Donoghue, A. C.; Hollfelder, F.; J. Am. Chem. Soc 2008, 130, 16547.
- 7. Zalatan, J. G.; Herschlag, D.; J. Am. Chem. Soc. 2006, 128, 1293.
- 8. Hemrika, W.; Renirie, R.; Dekker, H. L.; Barnett, P.; Wever, R.; Proc. Natl. Acad. Sci. USA 1997, 94, 2145.
- 9. Yoneyama, T.; Shiozawa, M.; Nakamura, M.; Suzuki, T.; Sagane, Y. Katoh, Y.; Watanabe, T.; Ohyama, T.; J. Biol. Chem. 2004, 279, 37477.
- 10. Bozzo, G. G.; Raghothama, K. G.;. Plaxton, W. C.; Eur. J. Biochem. 2002, 269, 6278.
- 11. Kenton, P.; Mur, L. A. J.; Draper, J.; J. Exp. Bot. 1999, 50, 1331.
- 12. Jakobek, J. L.; Lindgren, P. B.; J. Exp. Bot. 2002, 53, 387.
- 13. Oliveira, M. C. B.; Scarpellini, M.; Neves, A.; Terenzi, H.; Bortoluzzi, A. J.; Szpoganicz, B.; Greatti, A.; Mangrich, A. S.; Souza, E. M.; Fernandez, P. M.; Soares, M.; Inorg. Chem. 2005, 44, 921.
- 14. Rey, N. A.; Neves, A.; Bortoluzzi, A. J.; Pich, C. T.; Terenzi, H.; Inorg. Chem. 2007, 46, 348.
- 15. Deck, K. M.; Tseng, T. A.; Burstyn, J. N.; Inorg. Chem. 2002, 41, 669.
- 16. Hegg, E. L.; Burstyn, J. N.; J. Am. Chem. Soc. 1995, 117, 7015.
- 17. Hegg, E. L.; Burstyn, J. N.; Inorg. Chem. 1996, 35, 7474.
- 18. Parrilha, G. L.; Fernandes, C.; Bortoluzzi, A. J.; Szpoganicz, B.; Silva, M. S.; Pich, C. T.; Terenzi, H.; Horn, A., Jr.; Inorg. Chem. Commun. 2008, 11, 643.
- 19. Mitic, N.; Smith, S. J.; Neves, A.; Guddat, L. W.; Gahan, L. R.; Schenk, G.; Chem. Rev. 2006, 106, 3338.
- 20. Gavrilova, A. L.; Bosnich, B.; Chem. Rev. 2004, 104, 349;
- 21. Gahan, L. R.; Smith, S. J.; Neves, A.; Schenk, G.; Eur. J. Inorg. Chem. 2009, 2745.
- 22. Neves, A.; Lanznaster, M.; Bortoluzzi, A. J.; Peralta, R. A.; Casellato, A.; Castellano, E. E.; Herrald, P.; Riley, M. J.; Schenk, G.; J. Am. Chem. Soc. 2007, 129, 7486.
- 23. Karsten, P.; Neves, A.; Bortoluzzi, A. J.; Drago, V.; Lanznaster, M.; Inorg. Chem. 2002, 41, 4624.
- 24. Karsten, P.; Neves, A.; Bortoluzzi, A. J.; Strähle, J.; Maichle-Mössmer, C.; Inorg. Chem. Commun. 2002, 5, 434.
- 25. Batista, S. C.; Neves, A.; Bortoluzzi, A. J.; Vencato, I.; Peralta, R. A.; Szpoganicz, B.; Aires, V. E.; Severino, P. A.; Inorg. Chem. Commun. 2003, 6, 1161.
- 26. Lanznaster, M.; Neves, A.; Bortoluzzi, A. J.; Aires, V. V. E.; Szpoganicz, B.; Terenzi, H.; Severino, R. C.; Fuller, J. M.; Drew, S. C.; Gahan, L. R.; Hanson, G. R.; Riley, M. J.; Schenk, G.; J. Biol. Inorg. Chem. 2005, 10, 319.
- 27. Schenk, G.; Peralta, R. A.; Batista, S. C.; Bortoluzzi, A. J.; Szpoganicz, B.; Dick, A. K; Herrald, P.; Hanson, G. R.; Szilagyi, R. K.; Riley, M. J.; Gahan, L. R.; Neves, A.; J. Biol. Inorg. Chem. 2008, 13, 139.
- 28. Smith, S. J.; Casellato, A.; Hadler, K. S.; Mitić, N.; Riley, M. J.; Bortoluzzi, A. J.; Szpoganicz, B.; Schenck, G.; Neves, A.; Gahan, L.; J. Biol. Inorg. Chem. 2007, 12, 1207.
- 29. Neves, A.; Brito, M. A.; Drago, V.; Griesar, K.; Haase, W.; Inorg. Chim. Acta 1995, 237, 131.
- 30. Gonçalves, N. S.; Horn Jr., A.; Lanznaster, M.; Noda, L. K.; Neves, A.; J. Braz. Chem. Soc. 2006, 17, 1658.
- 31. Smith, S. J.; Jovito, R.; Horn Jr., A.; Nascimento, O. R.; Noble, C. J.; Hanson, G. H.; Stranger, R.; Jayaratne, V.; Cavigliasso, G.; Gahan, L. R.; Schenk, G.; Neves, A.; Riley, M. J.; unpublished results.
- 32. de Oliveira, I. R.W. Z.; Neves, A.; Vieira, I. C.; Sens. Actuators 2008, B 129, 424.
- 33. Bernhardt, P. V.; Schenk, G.; Wilson, G.; J. Biochem. 2004, 43, 10387.
- 34. Wang, D. L.; Holz, R. C.; David, S. S.; Que Jr., L.; Stankovich, M. T.; Biochemistry 1991, 30, 8187.
- 35. Bunton, C. A.; Farber, S. J.; J. Org. Chem 1969, 34, 767.
- 36. Martell, A. E.; Motekaitis, R. J.; Determination of Stability Constants, 2nd ed., VHC Publishers: Weinheim, Germany, 1992.
- 37. Enraf-Nonius; CAD-4 EXPRESS; Version 5.1/1.2; Enraf-Nonius: Delft, The Netherlands, 1994.
- 38. Spek, A.L.; HELENA; CAD-4 Data Reduction Program; Univ. of Utrecht, The Netherlands, 1996.
- 39. Spek, A.L.; Acta Crystallogr. 1990, A46, C34.
- 40. North, A. C. T.; Phillips, D. C.; Mathews, F. S.; Acta Crystallogr. 1968, A24, 351.
- 41. Altomare, A.; Burla, M. C.; Camalli, M.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R.; J. Appl. Cryst 1999, 32, 115.
- 42. Sheldrick, G.M.; Acta Crystallogr 2008, A64, 112.
- 43. Neves, A.; Rossi, L. M.; Bortoluzzi, A. J.; Szpoganicz, B.; Wiezbicki, C.; Schwingel Haase, E.; Ostrovsky, W. S.; Inorg. Chem. 2002, 41, 1788.
- 44. Peralta, R. A.; Neves, A.; Bortoluzzi, A. J.; dos Anjos, A.; Xavier, F. R.; Szpoganicz, B.; Terenzi, H.; de Oliveira, M. C. B.; Castellano, E.; Friedermann, G. R.; Mangrich, A. S.; Novak, M. A.; J. Inorg Biochem. 2006, 100, 992.
- 45. Neves, A.; Rossi, L. M.; Bortoluzzi, A. J.; Haase, W.; Werner. R.; J. Braz. Chem. Soc. 2001, 12, 747.
- 46. Gonzalez-Alvarez, M.; Alzuet, G.; Borras, J.; Macías, B.; Del Olmo, M.; Liu-Gonzalez, M. E.; Sanz, F.; J. Inorg. Biochem. 2002, 89, 29.
- 47. Scarpellini, M.; Neves, A.; Hörner, R.; Bortoluzzi, A. J.; Szpoganicz, B.; Zucco, C.; Nome, R. A.; Drago, V.; Mangrich. A.S.; Ortiz, W. A.; Passos, W.A.; de Oliveira, M. C.; Terenzi, H.; Inorg. Chem. 2003, 42, 8353.
- 48. Sreedhara, A.; Freed, J. D. E.; Cowan, J. A.; J. Am. Chem. Soc. 2000, 122, 8814.
- 49. Brezova, V.; Valko, M.; Breza, H.; Morris, H.; Telser, J.; Dvoranova, D.; Kaiserova, K.; Varecka, L.; Mazur, M.; Leibfritz, D.; J. Phys. Chem. B 2003, 107, 2415.
- 50. Macías, B.; Villa, M.V.; Gómez, B.; Borrás, J.; Alzuet, G.; González-Álvarez, M.; Castineiras, A.; J. Inorg. Biochem. 2007, 101, 444.
- 51. Uytterhoeven, K.; Sponer, J.; Van Meervelt, L.; Eur. J. Biochem. 2002, 269, 2868.
- 52. Lanznaster, M.; Neves, A.; Bortoluzzi, A. J.; Szpoganicz, B.; Schwingel, E.; Inorg. Chem 2002, 41, 5641.
- 53. Horn Jr, A.; Vencato, I.; Bortoluzzi, A.J.; Hörner, R.; Silva, R.A.N.; Szpoganicz, B.; Drago, V.; Terenzi, H.; Oliveira, M.C.B.; Werner, R.; Haase. W.; Neves, A.; Inorg. Chim. Acta 2005, 358, 339.
- 54. Bornscheuer, U. T.; Kazlauskas, R. J.; Angew. Chem. Int. Ed. 2004, 43, 6032.
- 55. Smith, S. J.; Noble, C. J.; Palmer, R. C.; Hanson, G. R.; Schenk, G.; Gahan, L. R.; Riley, M. J.; J. Biol. Inorg. Chem. 2008, 13, 499.
- 56. Cowan, J. A.; Curr. Opin. Chem. Biol. 2001, 5, 634.
- 57. Sreedhara, A.; Cowan, J. A.; J. Biol. Inorg. Chem. 2001, 6, 337.
Publication Dates
-
Publication in this collection
30 July 2010 -
Date of issue
2010
History
-
Received
01 Oct 2009 -
Accepted
15 Mar 2010