Abstract
The use of hydroxyurea (HU) can improve the clinical course of sickle cell disease. However, several features of HU treatment remain unclear, including the predictability of drug response and determination of adequate doses, considering positive responses and minimal side effects. In order to identify adequate doses of HU for treatment of sickle cell disease, 10 patients, 8 with sickle cell anemia and 2 with Sß thalassemia (8SS, 2Sß), were studied for a period of 6 to 19 months in an open label dose escalation trial (10 to 20 mg kg-1 day-1). Hemoglobin (Hb), fetal hemoglobin (Hb F) and mean corpuscular volume (MCV) values and reticulocyte, neutrophil and platelet counts were performed every two weeks during the increase of the HU dose and every 4 weeks when the maximum HU dose was established. Reduction in the number of vasoocclusive episodes was also considered in order to evaluate the efficiency of the treatment. The final Hb and Hb F concentrations, and MCV values were significantly higher than the initial values, while the final reticulocyte and neutrophil counts were significantly lower. There was an improvement in the concentration of Hb (range: 0.7-2.0 g/dl) at 15 mg HU kg-1 day-1, but this concentration did not increase significantly when the HU dose was raised to 20 mg kg-1 day-1. The concentration of Hb F increased significantly (range: 1.0-18.1%) when 15 mg HU was used, and continued to increase when the dose was raised to 20 mg kg-1 day-1. The final MCV values increased 11-28 fl (femtoliters). However, reticulocyte (range: 51-205 x 109/l) and neutrophil counts (range: 9.5-1.3 x 109/l) obtained at this dose were significantly lower than those obtained with 15 mg kg-1 day-1. All patients reported a decrease in frequency or severity of vasoocclusive episodes. These results suggest that a hydroxyurea dose of 15 mg kg-1 day-1 seems to be adequate for treatment of sickle cell disease in view of the minimal side effects observed and the improvement in laboratory and clinical parameters.
sickle cell disease; hydroxyurea; hemoglobinopathy
Braz J Med Biol Res, August 1997, Volume 30(8) 933-940
Departamento de Clínica Médica, Faculdade de Ciências Médicas, Universidade Estadual de Campinas, 13081-970 Campinas, SP, Brasil
 Patients and Methods
Patients and Methods
References
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes

The use of hydroxyurea (HU) can improve the clinical course of sickle cell disease. However, several features of HU treatment remain unclear, including the predictability of drug response and determination of adequate doses, considering positive responses and minimal side effects. In order to identify adequate doses of HU for treatment of sickle cell disease, 10 patients, 8 with sickle cell anemia and 2 with Sß thalassemia (8SS, 2Sß), were studied for a period of 6 to 19 months in an open label dose escalation trial (10 to 20 mg kg-1 day-1). Hemoglobin (Hb), fetal hemoglobin (Hb F) and mean corpuscular volume (MCV) values and reticulocyte, neutrophil and platelet counts were performed every two weeks during the increase of the HU dose and every 4 weeks when the maximum HU dose was established. Reduction in the number of vasoocclusive episodes was also considered in order to evaluate the efficiency of the treatment. The final Hb and Hb F concentrations, and MCV values were significantly higher than the initial values, while the final reticulocyte and neutrophil counts were significantly lower. There was an improvement in the concentration of Hb (range: 0.7-2.0 g/dl) at 15 mg HU kg-1 day-1, but this concentration did not increase significantly when the HU dose was raised to 20 mg kg-1 day-1. The concentration of Hb F increased significantly (range: 1.0-18.1%) when 15 mg HU was used, and continued to increase when the dose was raised to 20 mg kg-1 day-1. The final MCV values increased 11-28 fl (femtoliters). However, reticulocyte (range: 51-205 x 109/l) and neutrophil counts (range: 9.5-1.3 x 109/l) obtained at this dose were significantly lower than those obtained with 15 mg kg-1 day-1. All patients reported a decrease in frequency or severity of vasoocclusive episodes. These results suggest that a hydroxyurea dose of 15 mg kg-1 day-1 seems to be adequate for treatment of sickle cell disease in view of the minimal side effects observed and the improvement in laboratory and clinical parameters.
Key words: sickle cell disease, hydroxyurea, hemoglobinopathy
Abstract
Introduction
Sickle cell disease is the most common severe hemoglobinopathy. Despite the years of intensive research, there is no effective primary therapy available for these patients, who experience all the problems associated with a painful and chronic disease. For most patients, treatment is limited to a supportive environment, management of complications and pain crises, and to the detection of signs which may predict early morbidity or mortality. The development of primary therapies for sickle cell disease has received special attention, particularly those involved in the prevention or reversal of the polymerization of sickle hemoglobin (Hb S) in erythrocytes (1-3). The most encouraging approach to prevent Hb S polymerization has been the use of pharmacological agents which increase the production of fetal hemoglobin (Hb F) (1-6). Hydroxyurea (HU) induces Hb F synthesis (1,4-12) and can increase hemoglobin (Hb) concentration and mean corpuscular volume (MCV) in sickle cell disease (1,4,5,7,13,14). A decrease in frequency and severity of vasoocclusive episodes has been reported, suggesting a less severe course of the disease during drug administration (4,5,7,12).
However, many questions remain unanswered, such as which factors determine the increase in Hb and Hb F concentrations and in MCV values, whether these findings will eventually lead to a significant clinical improvement and which doses of HU are adequate for each patient, where positive responses and minimal side effects are considered. The range of optimal HU doses for obtaining the desired hematological and clinical effects is not known since most studies employed the maximum doses of HU tolerated.
In order to evaluate the adequacy of low doses of HU in the treatment of sickle cell disease, an open label dose escalation trial was undertaken, and three different doses of HU (10, 15, and 20 mg kg-1 day-1) were compared.
Patients and Methods
The following protocol was approved by the Hospital Ethics Committee and oral informed consent was obtained from the patients.
Eligibility requirements
Ten adult patients (18 years old or more) with sickle cell anemia or Sß thalassemia (8SS and 2Sß) seen at the University Hospital of Campinas and fulfilling one or more of the following criteria were eligible for the study: 1) at least three severe painful crises during the year before entering the study (including emergency room visits), 2) Hb concentrations lower than 6 g/dl, 3) prolonged priapism episodes (for more than 12 h), 4) previous cerebral vascular accident (CVA) and alloimmunization, and 5) chronic extensive leg ulcers. Two of the patients had had a stroke during childhood and had been receiving transfusion for at least ten years. Both developed alloimmunization and for one of them access to the vein for transfusion became difficult. The diagnosis of sickle cell disease was confirmed by hemoglobin electrophoresis performed on an acetate cellulose membrane, pH 8.6, and citrate agar gel, pH 6.2, and in most cases by family studies. Hb A2 was eluted for quantification (15). Hb F concentration was determined using the method of Singer with modifications (16). Globin gene haplotypes were determined as described elsewhere (17). Patients with aspartate aminotransferase (AST) or alanine aminotransferase (ALT) higher than 100 IU/l and creatinine higher than 1 mg/dl or creatinine clearance lower than 100 ml min-1 1.73 m-2 were excluded from the study.
Drug doses
The initial dose of HU (Bristol, Regensburg, Germany) was 10 mg kg-1 day-1, given once a day, and was increased by 5 mg kg-1 day-1 every 8 weeks, unless toxicity was present, to a maximum dose of 20 mg kg-1 day-1. Toxicity was defined by the presence of at least one of the following characteristics: reticulocytes <50 x 109/l, neutrophils <2 x 109/l, platelets <100 x 109/l, a 20% decrease in Hb concentration or an absolute value of less than 4.5 g/dl, a 50% increase in serum creatinine or a 100% increase in AST or ALT.
Laboratory studies
Basal peripheral blood cell counts were obtained at the beginning of the study, every 2 weeks during the increase of the drug dosage and every 4 weeks after the maximum HU dose was established. Hb concentration, MCV values and platelet counts were obtained with an automated instrument (Cell-Dyn, Model 1600 CS). The neutrophil and reticulocyte counts were obtained manually. AST and ALT were determined using a Boehringer kit (Mannheim, Germany) and a Merck kit (Darmstadt, Germany) was used for the determination of urea and creatinine values. All these parameters were measured every 4 weeks and the mean values were determined for different HU doses (10, 15 and 20 mg kg-1 day-1). Hb F concentrations were also evaluated according to gender and haplotype of sickle cell disease.
Clinical parameters
The frequency of severe painful crises for each patient was determined during the administration of HU and during an equal period immediately before HU administration and is reported as annual frequency computed by dividing the number of crises by the number of years elapsed. A crisis was defined as a painful episode lasting longer than 24 h. Frequencies of CVA, aseptic necrosis and priapism were also determined before and during HU administration. The diameters of leg ulcers were evaluated every month throughout the study.
Statistical analysis
The paired t-test or Wilcoxon test was used to compare the differences between initial and final measurements and the differences between the mean values obtained for different HU doses (18,19), depending on sample distribution. The Spearman correlation coefficient was used to determine bivariate associations between defined variables (19).
Results
Patients
Eight patients with sickle cell anemia (five men and three women) and two patients with Sß thalassemia (one man and one woman) were enrolled in the study in December 1993. The globin gene haplotypes among sickle cell anemia patients were BEN/CAR in five patients, CAR/CAR in two, and BEN/BEN in only one, and among Sß thalassemia patients, BEN in one and CAR in the other (Table 1).
HU was introduced due to severe painful crises in two patients, Hb concentration lower than 6 g/dl in three, priapism in two, CVA in two patients, and extensive leg ulcers in five. Four of the patients fulfilled more than one of the criteria required to enter the study. The ages ranged from 20 to 44 years (mean ± SD: 24.0 ± 5.9; median: 21.0). Patients were treated for a period of 6 to 19 months (mean: 11.2 ± 6.8; median: 21.0). Four patients were treated for 12 months or longer, one for 8 months, two for 7 months and three for 6 months. The initial HU dose was 15 mg kg-1 day-1 for two patients with low weight (less than 50 kg). At the end of the study (October, 1995), the examination of HU doses revealed that five patients received the maximal doses (20 mg kg-1 day-1) and the remaining five patients received 15 mg kg-1 day-1 doses of HU (Table 2).
Changes in Hb, Hb F and MCV
The Hb concentrations increased significantly when compared with the initial values (P<0.01). The increase ranged from 0.7 to 2.0 g/dl (mean ± SD: 1.3 ± 0.5; median: 1.2). In one of these patients (patient No. 1), the Hb concentration repeatedly exceeded 12.5 g/dl during the administration of 20 mg kg-1 day-1 HU. Although no morbidity was associated with this high Hb concentration, the HU dose was decreased to 15 mg kg-1 day-1. Two patients were excluded from the analysis because they had received packed red cell transfusions at the beginning of the study. The difference between mean Hb concentrations obtained during the administration of 10 and 15 mg kg-1 day-1 HU (P<0.05) was significantly greater than the difference between mean Hb concentrations obtained during the administration of 15 and 20 mg kg-1 day-1 HU (P = 0.60). These results are shown in Figure 1.
In all patients the Hb F concentration increased significantly when compared with the initial value (P<0.01), ranging from 1.0 to 18.1% (mean ± SD: 7.7 ± 5.3; median: 7.0). The mean Hb F concentrations obtained during the administration of 15 mg kg-1 day-1 HU were significantly higher than those obtained during the administration of 10 mg kg-1 day-1 (P<0.01). However, they were significantly lower (P<0.01) than those obtained during administration of 20 mg kg-1 day-1 HU. These results are shown in Figure 1. The response of Hb F to HU was not associated with gender or ß-globin haplotype.
In eight of ten patients the final MCV values increased significantly when compared with the initial values (P = 0.01). This increase ranged from 11-28 fl (femtoliters) (mean ± SD: 17.4 ± 5.8; median: 17.0). The mean MCV values obtained during the administration of 15 mg kg-1 day-1 HU were significantly higher than those obtained during the administration of 10 mg kg-1 day-1 (P<0.01). However, they were significantly lower than those obtained during the administration of 20 mg kg-1 day-1 HU (P<0.01). These results are shown in Figure 1.
There was no correlation between the increase of Hb and Hb F (r = -0.26; P = 0.46) or the MCV values (r = -0.23; P = 0.53), while the increase in MCV and Hb F concentration (r = 0.67; P = 0.03) was highly correlated.
- Effect of different doses of hydroxyurea (HU) on mean values of hemoglobin (Hb), fetal hemoglobin (Hb F) and mean corpuscular volume (MCV) for 8 patients with sickle cell anemia (patients 1-8) and 2 patients with Sß thalassemia (patients 9 and 10).
Toxicity
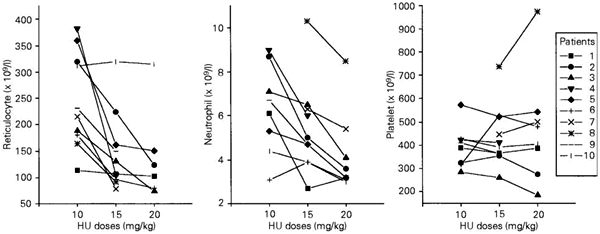
In all patients, the final reticulocyte counts were significantly lower than the initial ones (P<0.01). This decrease ranged from 51 x 109/l to 205 x 109/l (mean ± SD: 128.0 ± 70.0 x 109; median: 98.0 x 109). Since the reticulocyte count for one of the patients was performed after the beginning of the study, the patient was excluded from this analysis. The mean reticulocyte values obtained during the administration of 15 mg kg-1 day-1 HU were significantly lower than those obtained during the administration of 10 mg kg-1 day-1 (P<0.05). The same significant differences were observed between the values obtained during the administration of 20 and 15 mg kg-1 day-1 HU (P<0.05). These results are shown in Figure 2. The decrease in reticulocyte counts during treatment was not correlated with the increase in Hb concentration (r = 0.02; P = 0.095) or with the decrease in neutrophil counts (r = 0.031; P = 0.46).
The highest decrease of neutrophil counts was 9.5 x 109/l and the lowest decrease 1.3 x 109/l (mean ± SD: 3.8 ± 2.7; median: 3.0). In eight of ten patients the mean neutrophil values obtained during the administration of 15 mg kg-1 day-1 HU were lower than those obtained during the administration of 10 mg kg-1 day-1 (P<0.02). The final neutrophil counts were significantly lower than the initial values (P<0.01) but in two patients they were higher than or similar to the initial counts. Significant differences were also observed among values obtained during the administration of 20 and 15 mg kg-1 day-1 HU (P<0.01). These results are shown in Figure 2. Platelet counts did not change significantly during the study (Figure 2). No hematologic, hepatic or renal toxicity was observed in any of the ten patients.
- Effect of different doses of hydroxyurea (HU) on mean reticulocyte, neutrophil, and platelet counts for 8 patients with sickle cell anemia (patients 1-8) and 2 patients with Sß thalassemia (patients 9 and 10).
Clinical responses
The mean annual frequency of severe pain crises before HU administration was 1.3 (range: 0.0 to 6.0), and decreased to 0.6 (range: 0.0 to 4.0) during HU treatment. Although an improvement in frequency and severity of pain crises was reported by all patients, no significant differences in annual frequency were demonstrable, due to the small number of patients with pain crises enrolled in this study. However, one patient who presented moderate to severe crises at least every two months had no crisis during HU administration. During the study period there was no recurrence of CVA in the two patients who had suffered a previous stroke. There was a recurrence of priapism in one of the two patients who had priapism previously, but the second episode was not as severe as the first one. Reduction in the diameter of leg ulcers was not observed in any patient.
Hydroxyurea increases fetal hemoglobin production and improves the clinical course of sickle cell disease (1,4-12), but the adequate dose for each patient varies widely. In the present study, maximum Hb concentration was obtained in most patients with an intermediate dose of HU (15 mg kg-1 day-1). The Hb concentration obtained with this dose was significantly higher than that obtained with 10 mg kg-1 day-1. No significant difference was observed in Hb levels when the dose was raised to 20 mg kg-1 day-1. Although the number of patients was small, these results suggest that 15 mg kg-1 day-1 HU is sufficient to improve Hb concentration. Previous studies (5,7) determined the maximum doses of HU tolerated but not the minimum efficient dose as done in the present study. Moreover, to our knowledge, there are no studies on the use of HU in Brazilian patients with sickle cell disease.
During the increase of the dose of HU from 10 to 20 mg kg-1 day-1 there was an increase in Hb F concentration and a decrease in reticulocyte counts, as described previously (5,7,10-12). However, these findings did not significantly correlate with the increase in Hb concentration. Despite the risks of hemoglobin increase without a concomitant increase in fetal hemoglobin which could represent a higher risk of sickling, patient No. 1, who showed a concentration of Hb higher than 12.5 g/dl, did not have any vasoocclusive episode during the study. However, we decided to reduce his HU dose. It is well known that red cell survival increases during HU therapy (4,5,13,20).
In the present study, the increase in Hb F was not related to gender, probably due to the reduced number of patients enrolled, or to ß-globin haplotypes, since most patients were heterozygous for BEN/CAR haplotypes.
An evident result of HU treatment was the increase in MCV. Improvement in red cell diameter was correlated with the increase in HU dose, as observed previously (4,5,7). The increase in MCV during HU treatment is primarily due to an increase in cell Hb content but may also reflect altered properties of red cell membranes or the increase in the water content of red cells (2,7,14).
Although hydroxyurea has been used in patients with CVA, there are reports of recurrence and death when transfusion treatment is stopped and followed by treatment with hydroxyurea only (10). In the present study, we started HU treatment in two patients who presented one episode of stroke during childhood and who had been receiving transfusion treatment for at least twelve years. One of them was also receiving deferoxamine. The decision to start with HU in these cases was based on the fact that both patients had developed alloimmunization which prevented transfusion and one of them had a very difficult vein access. No recurrence of stroke occurred in either case during the study.
The frequency and severity of vasoocclusive crises appeared to decrease in all patients, in accordance with other reports (5,7,12). However, no significant differences in annual frequency were demonstrable, probably due to the reduced number of patients with pain crises.
Although a decrease in reticulocyte and neutrophil counts was observed in most of the patients, critical values were not reached in any of them. None of the patients presented hepatic or renal toxicity or an increase in infection frequency during HU treatment.
The present results suggest that a considerable proportion of patients obtained an improvement in Hb and Hb F concentration and MCV values during HU administration and that a 15 mg kg-1 day-1 dose of HU appeared to be adequate for sickle cell disease patients, considering positive responses and minimal side effects, and indicate that a larger number of patients should be evaluated using lower doses of HU.
Discussion
Acknowledgments
The authors are indebted to Eliani Guelli, Hélio José de Abreu and Lusane Leão Baia for assistance with the statistical analysis and to Rosana Aparecida Gon Rocha and Sandra Souza de Andrade for the art work.
Address for correspondence: S.T.O. Saad, Hemocentro, UNICAMP, Caixa Postal 6198, 13081-970 Campinas, SP, Brasil.
Research supported by CNPq and FAPESP. Received September 25, 1996. Accepted July 8, 1997.
- 1. Fibach E, Burke LP, Schechter AN, Nogusj CT & Rodgers GP (1993). Hydroxyurea increases fetal hemoglobin in cultured erythroid cells derived from normal individuals and patients with sickle cell anemia. Blood, 81: 1630-1635.
- 2. Platt OS (1995). Sickle cell paths converge on hydroxyurea. Nature Medicine, 1: 307-308.
- 3. Schechter AN & Rodgers GP (1995). Sickle cell anemia - basic research reaches the clinic. New England Journal of Medicine, 332: 1372-1374.
- 4. Dover GJ & Charache S (1992). Hydroxyurea in induction of fetal hemoglobin synthesis in sickle cell disease. Seminars in Oncology, 19: 61-66.
- 5. Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, McMahon RD & Bonds DRP (1995). Effect of hydroxyurea on the frequency of painful crisis in sickle cell anemia. New England Journal of Medicine, 332: 1317-1322.
- 6. Eaton WA & Hofrichter J (1995). The biophysics of sickle cell hydroxyurea therapy. Science, 268: 1142-1143.
- 7. Charache S, Dover GJ, Moore RD, Eckert S, Ballas SK, Koshy M, Milner PFA, Orringer EP & Phillips Jr J (1992). Effects of hemoglobin F production in patients with sickle cell anemia. Blood, 79: 2555-2565.
- 8. Horiuchi K, Stephens MJ, Adachi K, Asakura T, Schwartz E & Ohene-Frempong K (1993). Image analysis studies of the degree of irreversible deformation of sickle cells in relation to cell density and Hb F level. British Journal of Haematology, 85: 356-364.
- 9. Rodgers GP, Olivieri NF, Sullivan K & Kan YW (1994). Current and future strategies for the management of hemoglobinopathies and thalassemia. In: McArthur JR & Benz EJ (Editors), Educational Program of the American Society of Hematology, Hematology-1994 Copyright, The American Society of Hematology, Nashville, TN 9-19.
- 10. Vichinsky EP & Lubin BH (1994). A cautionary note regarding hydroxyurea in sickle cell disease. Blood, 83: 1124-1128.
- 11. Steinberg MH, Lu ZH, Barton FB, Terrin ML, Charache S & Dover GJ (1997). Fetal hemoglobin in sickle cell anemia: determinants of response to hydroxyurea. Multicenter Study of Hydroxyurea. Blood, 89: 1078-1088.
- 12. Dover GJ, Brusilow S & Charache S (1994). Induction of fetal hemoglobin production in subjects with sickle cell anemia by oral sodium phenylbutyrate. Blood, 84: 339-343.
- 13. Ballas SK, Dover GJ & Charache S (1989). Effect of hydroxyurea on the rheological properties of sickle erythrocytes "in vivo". American Journal of Hematology, 32: 104-111.
- 14. Orringer EP, Blythe DSB, Johnson AE, Philips G, Dover GJ & Parker JC (1991). Effects of hydroxyurea on hemoglobin F and water content in the red blood cells of dogs and of patients with sickle cell anemia. Blood, 78: 212-216.
- 15. Weatherall DJ & Clegg JG (1981). The Thalassemia Syndromes. 3rd edn. Blackwell Scientific Publications, Oxford.
- 16. Dacie JV & Lewis SM (1984). Practical Haematology 6th edn. Churchill Livingstone, Edinburgh.
- 17. Costa FF, Arruda VR, Gonçalves MG, Miranda SRP, Carvalho MH, Sonati MF, Saad STO, Gesteira F, Fernandes D, Nascimento ML & Queiroz IL (1994). ßS gene-cluster haplotypes in sickle cell anemia patients from two regions of Brazil. American Journal of Hematology, 45: 96-97.
- 18. Agresti A & Finlay B (1986). Statistical Methods for the Social Sciences 2nd edn. Dellen Publishing Company, San Francisco.
- 19. Conover WJ (1971). Practical Nonparametric Statistics John Wiley & Sons, New York.
- 20. Goldberg MA, Brugnara C, Dover GJ, Scharpira L, Charache S & Bunn HF (1990). Treatment of sickle cell anemia with hydroxyurea and erythropoietin. New England Journal of Medicine, 323: 366-372.
Minimal doses of hydroxyurea for sickle cell disease
Correspondence and Footnotes
Publication Dates
-
Publication in this collection
28 Nov 2001 -
Date of issue
Aug 1997
History
-
Accepted
08 July 1997 -
Received
25 Sept 1996