Resumo
The general methodology of classical trajectories as applied to elementary chemical reactions of the A+BC type is presented. The goal is to elucidate students about the main theoretical features and potentialities in applying this versatile method to calculate the dynamical properties of reactive systems. Only the methodology for two-dimensional (2D) case is described, from which the general theory for 3D follows straightforwardly. The adopted point of view is, as much as possible, that of allowing a direct translation of the concepts into a working program. An application to the reaction O(¹D)+H2->O+OH with relevance in atmospheric chemistry is also presented. The FORTRAN codes used are available through the web page <a href="http://www.qqesc.qui.uc.pt">www.qqesc.qui.uc.pt</a>.
classical trajectories; Monte Carlo sampling; cross sections
classical trajectories; Monte Carlo sampling; cross sections
EDUCAÇÃO
O método das trajectórias clássicas: colisões coplanares do tipo A+BC
Classical trajectory method: A+BC coplanar collisions
Jorge M. C. Marques; Antonio Riganelli; António J. C. Varandas* * E-mail: varandas@qtvs1.qui.uc.pt
Departamento de Química, Universidade de Coimbra, 3004-535 Coimbra, Portugal
ABSTRACT
The general methodology of classical trajectories as applied to elementary chemical reactions of the A+BC type is presented. The goal is to elucidate students about the main theoretical features and potentialities in applying this versatile method to calculate the dynamical properties of reactive systems. Only the methodology for two-dimensional (2D) case is described, from which the general theory for 3D follows straightforwardly. The adopted point of view is, as much as possible, that of allowing a direct translation of the concepts into a working program. An application to the reaction O(1D)+H2®O+OH with relevance in atmospheric chemistry is also presented. The FORTRAN codes used are available through the web page www.qqesc.qui.uc.pt.
Keywords: classical trajectories; Monte Carlo sampling; cross sections.
INTRODUÇÃO
O estudo teórico de reacções químicas elementares em fase gasosa (como, por exemplo, reacções do tipo A+BC) envolve, em geral, três etapas fundamentais: (i) cálculo do potencial electrónico, sob acção do qual os núcleos se movem; (ii) resolução das equações que descrevem o movimento dos núcleos para um dado conjunto de condições iniciais e cálculo dos parâmetros microscópicos (e.g., secções eficazes); (iii) cálculo dos coeficientes cinéticos, com base na mecânica estatística e nos parâmetros microscópicos obtidos na etapa anterior. As duas primeiras etapas resultam da aproximação de Born-Oppenheimer1, segundo a qual é possível, em determinadas circunstâncias, tratar separadamente os movimentos electrónico e nuclear. Contudo, existem situações de forte acoplamento entre o movimento electrónico e o movimento nuclear, onde a aproximação de Born-Oppenheimer já não é válida2. Por seu lado, a etapa (iii) assume que é possível tratar separadamente os problemas dinâmico e estatístico, i.e., o tempo de interacção das moléculas que colidem é muito inferior ao tempo que decorre entre colisões subsequentes (fase gasosa diluída).
Neste trabalho concentramo-nos na resolução das equações do movimento nuclear. É evidente que um processo atómico-molecular é intrinsecamente de natureza quântica. Todavia, a aplicação da mecânica quântica ao estudo de uma reacção não constitui tarefa fácil e a sua utilização rotineira é ainda impossível. Uma alternativa ao uso de métodos quânticos consiste em assumir como válida a mecânica clássica para descrever o movimento dos núcleos. Formalmente, as trajectórias clássicas constituem o limite dos processos de espalhamento mecânico-quântico para energias e massas elevadas, i.e., em que se possa considerar um contínuo de estados. De facto, a experiência adquirida do estudo teórico de reacções químicas elementares tem mostrado que os métodos clássicos produzem resultados em boa concordância com os obtidos a partir da mecânica quântica mesmo em situações extremas como é o caso da bem conhecida reacção H+H2®H2+H3,4.
Na formulação de Hamilton, as equações clássicas do movimento dos núcleos assumem forma:

onde qi e pi, funções do tempo t, representam as coordenadas generalizadas e os correspondentes momentos conjugados associados aos ƒ graus de liberdade do sistema, respectivamente; H é o Hamiltoniano (clássico) dado por

sendo T e V as energias cinética e potencial do sistema, respectivamente. Assim sendo, considera-se que o sistema descreve uma trajectória no espaço de fase (qi, pi), a qual é determinada à partida pelo conjunto das condições que caracterizam o seu estado dinâmico no instante inicial (t=0). No caso particular de uma colisão molecular, as condições iniciais ficam perfeitamente estabelecidas quando se definem os parâmetros de colisão, i.e., as variáveis geométricas e energéticas que a caracterizam.
É evidente que a simulação de uma reacção química elementar não pode ser feita apenas com uma única trajectória, mas surge como o resultado da análise de um conjunto (em geral, grande) de trajectórias caracterizadas por diferentes valores das condições iniciais. Assim, a probabilidade de reacção (Pr) para um estado energético específico inicial é o resultado de uma média pesada sobre todos os parâmetros de colisão (c)5,o que se pode traduzir através do integral multidimensional

onde dc é o elemento de volume associado ao conjunto das variáveis de integração; C é uma constante de normalização e  é a função probabilidade para o conjunto c de parâmetros de colisão que definem a geometria inicial do sistema. (Note-se que só tem dois valores possíveis: 1 ou 0, conforme o conjunto particular de parâmetros de colisão conduz ao evento reactivo pretendido ou não.) Uma vez obtido o valor de Pr podem então calcular-se os parâmetros dinâmicos e cinéticos da reacção.
é a função probabilidade para o conjunto c de parâmetros de colisão que definem a geometria inicial do sistema. (Note-se que só tem dois valores possíveis: 1 ou 0, conforme o conjunto particular de parâmetros de colisão conduz ao evento reactivo pretendido ou não.) Uma vez obtido o valor de Pr podem então calcular-se os parâmetros dinâmicos e cinéticos da reacção.
O método das trajectórias clássicas (muitas vezes identificado pelo acrónimo QCT, do inglês quasiclassical trajectory) teve um grande desenvolvimento e aplicação nas últimas quatro décadas5-17, devido essencialmente ao aparecimento dos computadores digitais. Estes possibilitaram, por um lado, o cálculo massivo de trajectórias mesmo para reacções químicas envolvendo sistemas com muitos átomos, por outro, a divulgação do método QCT nomeadamente através da construção de códigos standard que têm contribuído para a sua utilização generalizada por parte da comunidade científica. Na Tabela 1 apresentam-se alguns dos programas de trajectórias mais usados. Contudo, a utilização destes programas em cursos de graduação em Química não são muito aconselháveis, dado que o seu tamanho e complexidade dificultam, a este nível, o entendimento da metodologia envolvida. Neste sentido, foram publicados vários trabalhos18-20 de índole educacional que consideram a aplicação do método QCT a casos mais simples.
O principal objectivo deste trabalho é o da apresentação detalhada do método QCT para o caso 2D, permitindo ao estudante construir o seu próprio programa de computador. Este artigo foi, pois, elaborado tendo em vista a escrita do programa traj2d.f que disponibilizamos no site www.qqesc.qui.uc.pt. Na próxima secção apresenta-se a metodologia envolvida no cálculo de trajectórias clássicas, enquanto na secção seguinte é discutida a obtenção de secções eficazes de reacção e diferencial. De seguida, aplica-se a metodologia descrita ao estudo da reacção O(1D)+H2®OH+H. Finalmente, a última secção sumaria algumas conclusões.
METODOLOGIA
A metodologia associada a um cálculo de trajectórias aplica-se aqui ao caso de reacções do tipo A+BC em 2D, sendo a sua extensão para 3D relativamente imediata embora um pouco mais complicada; aconselha-se ao leitor a consulta das Refs. 5,9. Tal metodologia encontra-se implementada no programa traj2d.f escrito em FORTRAN 77 pelos presentes autores.
Generalidades
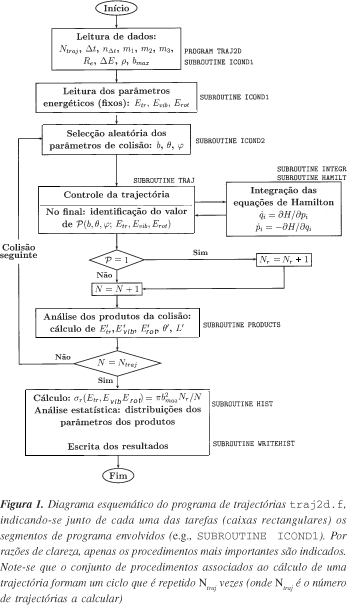
A Figura 1 apresenta um esquema geral dos procedimentos utilizados num cálculo de trajectórias para uma energia total fixa, caracterizada pelas suas componentes translacional (Etr), vibracional (Evib), rotacional (Erot) e potencial. Note-se que junto dos vários procedimentos se indicam os segmentos do programa traj2d.f em que eles se encontram implementados. Genericamente, e de acordo com o diagrama da Figura 1, um cálculo de trajectórias pode dividir-se em 5 etapas fundamentais: (i) leitura dos dados, (ii) selecção aleatória dos parâmetros de colisão e estabelecimento das condições iniciais, (iii) integração das equações de Hamilton, (iv) análise dos produtos da colisão (reactiva ou não) e, após o cálculo de um número pré-estabelecido de trajectórias (Ntraj), (v) análise estatística de todas as colisões (incluindo o cálculo da secção eficaz de reacção e as distribuições dos parâmetros dos produtos) e escrita dos resultados. Além disso, existem ainda outras tarefas auxiliares implementadas no programa traj2d.f que são importantes nomeadamente na etapa (ii). Contudo, por razões de clareza, não são indicadas explicitamente na Figura 1, mas serão igualmente abordadas nas subsecções seguintes.
O programa traj2d.f permite ainda escrever, para Ntraj = 1, todos os parâmetros dinâmicos relevantes ao longo de uma trajectória. A escrita dessas variáveis (e.g., coordenadas, momentos, energias em função do tempo) é feita no segmento de programa que controla a evolução da trajectória. Tal informação permite, entre outras coisas, verificar a conservação da energia total ao longo da trajectória e efectuar a representação gráfica das distâncias internucleares em função do tempo, o que constitui um dado importante sobre o tipo de colisão envolvida (além do seu valor ilustrativo).
Coordenadas
As posições dos três átomos envolvidos numa colisão do tipo A+BC são definidos no espaço Cartesiano através de um total de 9 coordenadas (6 coordenadas em 2D). Estas podem, em princípio, ser utilizadas como coordenadas generalizadas nas equações de Hamilton [Equações (1) e (2)]. No entanto, é frequente adoptar as coordenadas de Jacobi, pois estas permitem reduzir o número de equações de Hamilton a integrar, como veremos mais à frente. Em 2D, as coordenadas de Jacobi definem-se do seguinte modo: 2 coordenadas (q1, q2) definem a posição do átomo C em relação a B; 2 outras coordenadas (Q1, Q2) estabelecem a posição do centro de massa da molécula BC relativamente ao átomo A; finalmente, 2 coordenadas (Q1, Q2) definem o centro de massa do sistema formado pelos três átomos. Na Figura 2 mostra-se a relação entre as coordenadas de Jacobi e as coordenadas cartesianas dos átomos A, B, e C, i.e., (XA, YA), (XB, YB), e (XC, YC). Então, em termos das coordenadas XA, XB, e XC, essa relação exprime-se sob a seguinte forma matricial:

onde, para utilização futura, a matriz transformação é designada por A. Por seu lado, as restantes coordenadas de Jacobi q2, Q2, e Q2 estão associadas à direcção Y segundo uma equação idêntica à Equação (5). é evidente que a matriz inversa de A (i.e., A-1) permite obter as coordenadas (XA, XB, XC) a partir das coordenadas (q1, Q1, Q1), de acordo com a equação:

Esta relação é útil para o cálculo das coordenadas internucleares R1, R2, e R3 que, assim, podem ser facilmente obtidas a partir das coordenadas de Jacobi, usando as expressões

Equações de Hamilton
O Hamiltoniano do sistema [Equação (3)] pode ser explicitado em termos de coordenadas de Jacobi do seguinte modo:

onde pi, Pi, e Pi são os momentos conjugados das coordenadas qi, Qi, e Qi, respectivamente; as massas reduzidas µBC=mBmC/( mB+mC) e µA,BC=mA(mB+ mC)/M (onde M=mA+mB+mC) surgem naturalmente associadas na Equação (10) aos momentos relativos pi e Pi, respectivamente. Dado não existirem forças externas a actuar no sistema, o potencial electrónico V (previamente obtido) só depende das posições relativas dos núcleos (em geral, é função de R1, R2, e R3), pelo que só depende das coordenadas (relativas) qi e Qi (i=1, 2); ver Equações (7)-(9). Logo, tanto Pi como Qi são constantes do movimento, anulando-se as correspondentes equações de Hamilton, i.e.:

Para anular a Equação (12) foi necessário considerar que o centro de massa do sistema não se move (i.e., Pi = 0), o que se justifica pelo facto de Pi ser uma constante do movimento [cƒ. Equação (11)]. Assim, o número de equações do movimento [i.e., Equações (1) e (2)] a integrar pode ser reduzido de 12 para 8. Dado o Hamiltoniano da Equação (10), as 8 equações clássicas do movimento podem ser escritas explicitamente como

Nas Equações (15) e (16), o segundo factor (entre parênteses recto) de cada parcela constitui a derivada parcial da coordenada Rj (j=1-3) em ordem à respectiva coordenada de Jacobi e resulta da aplicação da regra da cadeia, i.e., 
Na literatura encontram-se inúmeros métodos21 que permitem fazer a integração numérica das Equações (13)-(16), sempre que não exista uma solução analítica (o que constitui a regra para a maior parte das aplicações com interesse em Química). Um método simples e de fácil implementação é o método de Runge-Kutta de quarta ordem.
Condições iniciais
A determinação de uma solução particular para um conjunto de equações diferenciais acopladas de primeira ordem requer o conhecimento do ponto de partida para a integração, i.e., o estabelecimento das condições iniciais do problema. No caso das equações clássicas do movimento, essas condições definem o estado dinâmico inicial do sistema; para uma colisão do tipo A+BC em 2D terão de ser definidas 4 coordenadas e 4 momentos iniciais.
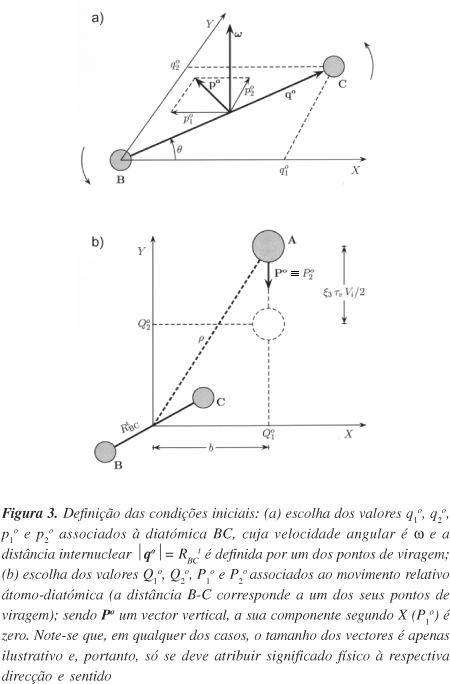
É evidente que o modo como é escolhido o conjunto inicial de coordenadas e momentos com vista à integração das equações de Hamilton depende do tipo de análise estatística que se pretende fazer. Como vimos na Introdução, para um estado energético inicial caracterizado pelos valores das energias de colisão (Etr), vibracional (Evib), e rotacional (Erot), a probabilidade de reacção [Equação (4)] obtém-se por integração sobre todos os parâmetros de colisão que, para A+BC em 2D, se encontram definidos na Figura 3, i.e., parâmetro de impacto (b), ângulo de colisão (q) e a fase de vibração (j) da diatómica BC. Assim, o integral da Equação (4) pode ser reescrito sob a forma

onde se indica, entre parênteses, a função de distribuição normalizada para cada uma das variáveis de colisão b, q e j; bmax é o máximo valor do parâmetro de impacto para o qual deixa de haver reacção e é, habitualmente, calculado através do método tentativa/erro para um número restrito de trajectórias. O cálculo do integral multidimensional da Equação (17) é geralmente conseguido com o recurso ao método de Monte Carlo, onde se procede a uma escolha aleatória das variáveis de integração. Tal procedimento5 deverá conduzir a uma distribuição uniforme segundo todas as dimensões do integral da Equação (17). Isto quer dizer que, representando dƒ=2b db/bmax2, dg=dq /2p e dh=dj/2p as distribuições normalizadas das variáveis de colisão, terá então de se obter uma amostragem uniforme das funções integrais ƒ(b) = b2/bmax2 , g(q)=q/2p e h(j)=j/2p no intervalo [0, 1]. (Note-se que para o parâmetro de impacto a distribuição é uniforme em b2.) Assim, as variáveis de colisão são seleccionadas segundo as expressões:

onde x1, x2 e x3 são três números aleatórios seleccionados no intervalo [0, 1].
Na Figura 3 mostra-se um modo de definir, a partir dos parâmetros de colisão, as coordenadas iniciais qio e respectivos momentos conjugados pio associados à diatómica reagente BC [painel (a)], e também as coordenadas iniciais átomo-diatómica Qio, bem como os seus momentos conjugados Pio [painel (b)]. É fácil concluir da Figura 3 que um modo de simular a fase de vibração da molécula BC, equivalente ao da Equação (20), surge através da escolha aleatória da coordenada Q2o no intervalo [(r2-b2)1/2 , (r2-b2)1/2- tvVi/2]. Note-se que r é a distância inicial entre os dois fragmentos (A e BC) e tvVi/2 é a distância percorrida pelo átomo A, cuja velocidade inicial é Vi [sendo Vi = (2µA,BCEtr)1/2 ], em meio período de vibração tv/2 da molécula diatómica BC. De facto, é habitual fixar a molécula diatómica num dos seus pontos de viragem (i.e., metade das trajectórias começam no ponto de viragem interno Rint, enquanto a outra metade parte do externo Rext), dado que nesse caso a velocidade segundo o eixo internuclear de BC é zero. Então, de acordo com a Figura 3 obtemos o valores iniciais das 4 coordenadas:

onde RBCt coincide alternadamente com os pontos de viragem Rint e Rext, sendo estes calculados (antes da integração da primeira trajectória) por resolução numérica da equação:

onde V(R2) representa a função energia potencial para a diatómica BC. Note-se que a Equação (25) assume a separabilidade dos movimentos de vibração e rotação, sendo o valor (constante) do momento angular rotacional obtido, para a distância de equilíbrio (Re) da molécula diatómica BC, segundo a expressão habitual

Por seu lado, tv /2 pode ser obtido por integração numérica das equações de Hamilton para o movimento vibro-rotacional da diatómica BC entre os pontos de viragem interno e externo.
Uma vez estabelecidas as coordenadas iniciais, resta agora definir os seus momentos conjugados. Para isso, devemos ter em conta as diferentes contribuições para a energia total do sistema: translacional, vibracional e rotacional. A contribuição vibracional é zero uma vez que a diatómica parte de um dos seus pontos de viragem. Por outro lado, de acordo com a escolha feita para a direcção da velocidade inicial de colisão, a contribuição translacional só tem componente segundo o eixo Y. Quanto à contribuição rotacional deve notar-se que, em 2D, BC só pode rodar no plano, sendo a velocidade angular w (e, portanto, o momento angular rotacional) perpendicular a esse plano. Então, as componentes, p1o e p2o, do momento po são perpendiculares a w, podendo ser calculadas usando a expressão

onde o sinal ´ designa o produto vectorial entre w e o vector posição inicial do átomo C relativamente ao B (qo); ver Figura 3(a). Sendo estes dois vectores mutuamente perpendiculares, os momentos iniciais podem então obter-se recorrendo à Equação (27):

onde o valor da velocidade angular em cada ponto de viragem pode ser obtido pela expressão

Assim, dado o estado inicial (Etr, Evib, Erot) e os valores das variáveis r, bmax, obtemos os valores iniciais das coordenadas e dos respectivos momentos conjugados.
Análise dos produtos da colisão
Uma trajectória termina sempre que duas ou mais distâncias internucleares excederem um valor pré-definido. É habitual definir esse valor igual à distância átomo-diátomo inicial (r).
Após a colisão, para além de se pretender saber o tipo de produtos que se formam, é importante conhecer o seu estado interno (Etr', Evib', Erot') bem como o ângulo de espalhamento (i.e., o ângulo formado pelos vectores velocidade inicial e velocidade final). Para obter essas grandezas associadas aos produtos da colisão é necessário calcular novas coordenadas de Jacobi e novos momentos conjugados. Assim, vamos considerar na exposição que se segue que se formam os produtos C+AB (para os produtos B+AC o tipo de transformações seria em tudo semelhante e, por isso, é dispensável a apresentação). Na Figura 4 mostra-se o novo sistema de coordenadas de Jacobi (qi', Qi', Qi'), as quais se definem de modo idêntico ao dado pela Equação (5) para as coordenadas qi, Qi e Qi. Sob a forma matricial as coordenadas q1', Q1', e Q1' definem-se como

sendo a matriz transformação designada doravante por B; ver a matriz A da Equação (5). Do mesmo modo, as coordenadas q2', Q2', e Q2' definem-se usando a matriz B e as coordenadas YA, YB e YC.
É possível representar as coordenadas (q1', Q1', Q1') em termos das coordenadas originais (q1, Q1, Q1), atendendo a que o vector das coordenadas Cartesianas (YA, YB, YC) na Equação (33) se pode escrever como na Equação (6). Então, substituindo a Equação (6) na Equação (33), obtêm-se as novas coordenadas

onde a matriz transformação resulta do produto BA-1; ver também a Figura 4.
O Hamiltoniano do sistema em função das novas coordenadas e momentos é dado pela expressão:

onde µAB=mAmB/( mA+mB) e µC,AB=mC(mA+ mB)/M são as massas reduzidas átomo-átomo e átomo-diatómica, respectivamente. Com base neste Hamiltoniano e usando um pouco de álgebra (como se apresenta detalhadamente no Apêndice), é fácil obter os novos momentos pi', Pi' e Pi' em função dos momentos pi, Pi e Pi, nomeadamente

É claro da Figura 4 e das Equações (34) e (36), que as coordenadas do centro de massa e respectivos momentos conjugados não sofrem alteração ao mudar-se de sistema de coodenadas, não sendo portanto relevantes para a análise dos produtos de colisão.
Uma vez calculadas as novas coordenadas e os respectivos momentos conjugados, podem então obter-se as propriedades dinâmicas associadas aos produtos resultantes da colisão. Entre elas, consideramos o cálculo da energia translacional de afastamento do átomo C relativamente à diatómica AB,

e da energia interna da diatómica AB dos produtos

onde V(R1) representa a energia potencial de AB. Por seu lado, a energia total do sistema (Etot´) pode ser dada através da Equação (10) ou da Equação (35). Para obter a energia rotacional da diatómica BC usa-se a expressão habitual

onde o momento angular de rotação resulta do produto externo entre o vectores das coordenadas e dos momentos conjugados da diatómica AB:

Finalmente, a energia vibracional da diatómica AB obtém-se pela diferença entre a energia interna [Equação (38)] e a energia rotacional [Equação (39)]. Um outro parâmetro importante é o ângulo de espalhamento que é dado por

onde as componentes dos vectores velocidades inicial (Vi) e final (Vƒ) são dadas pelas equações:

Por convenção, o vector Vi diz respeito à velocidade relativa do átomo A que se aproxima, enquanto Vƒ é a velocidade relativa da diatómica AB dos produtos.
SECÇÕES EFICAZES E DISTRIBUIÇÕES
Já vimos na secção anterior que o cálculo da probabilidade de reacção para um dado estado inicial (Etr, Evib, Erot) se faz usando o método de Monte Carlo. Para isso, calcula-se um número (N) tão grande quanto possível de trajectórias, cujas condições iniciais são escolhidas de modo a cobrirem uniformemente o espaço de integração. Destas trajectórias contabilizam-se como reactivas (Nr) as que conduziram aos produtos pretendidos, sendo a probabilidade aproximada pela expressão

Note-se que esta expressão só será exacta no limite em que o número total de trajectórias tenda para infinito.
A secção eficaz de reacção é a área do círculo cujo raio é o parâmetro de impacto máximo, pesada com a probabilidade de reacção [Equação (46)], logo

O desvio padrão para um intervalo de confiança de 68 % é dado por14

A secção eficaz diferencial, por seu lado, estabelece a dependência da reactividade em relação ao ângulo de espalhamento (q´) e pode definir-se de acordo com a expressão7,22,23:

onde v representa as coordenadas angulares no referencial associado ao centro de massa do sistema e Nr(q') é o número de trajectórias reactivas para o ângulo de espalhamento q'; na prática, divide-se o intervalo [0, p] (i.e., intervalo a que pertence q') em subintervalos finitos Dq', para os quais se considera o número de trajectórias reactivas Nr. Note-se que o factor que aparece no denominador da segunda fracção resulta da distribuição angular ser independente do ângulo azimutal. Tal deve-se ao facto do processo de espalhamento ter simetria cilíndrica à volta do vector velocidade inicial24 e, assim, a distribuição angular nos produtos depende apenas do ângulo polar q'25. Logo,

onde o factor 2p resulta da integração sobre o ângulo azimutal.
Uma análise detalhada dos produtos da reacção A+BC pressupõe o cálculo das distribuições das energias translacional, vibracional e rotacional nos produtos que resultam do processo de colisão. Estas distribuições energéticas dão informação sobre as transferências de energia que ocorrem durante a reacção. Então, é habitual dividir o intervalo de energias em subintervalos, distribuindo as trajectórias reactivas por cada um deles (tal como explicámos atrás para o ângulo de espalhamento no cálculo da secção eficaz diferencial). Este processo, mais ou menos arbitrário, designa-se por histogramação, sendo especialmente utilizado quando se tem um contínuo de energias.
Um outro tipo de distribuição que dá informação relevante sobre o mecanismo de reacção é a função opacidade, i.e., probabilidade de reacção em função do parâmetro de impacto. Neste caso, a histogramação consiste em dividir o intervalo [0, bmax] em subintervalos, pelos quais se distribuem as trajectórias reactivas.
APLICAÇÃO À REACÇÃO O (1D)+H2
Escolheu-se a reacção O(1D)+H2®OH+H para aplicar o programa traj2d.f, a qual origina a espécie intermediária H2O, bem conhecida de qualquer estudante de Química. Acresce que existem26-31 vários potenciais que descrevem tal reacção. Embora não se dedique muito tempo a descrever a função potencial (funciona como uma espécie de "caixa negra"), optou-se por utilizar uma das mais simples funções potenciais disponíveis na literatura28,32. A Figura 5 ilustra detalhadamente a energética da reacção O(1D)+H2 para o potencial de Murrell e Carter28 utilizado neste trabalho. Note-se que o programa traj2d.f considera o zero do potencial nos reagentes, pelo que o mínimo profundo correspondente à espécie intermediária H2O se encontra a -167.4 kcal mol-1 do canal O(1D)+H2. Como conseqüência da reacção ser exoenergética (cf. DE=-42.2 kcal mol-1 na Figura 5), a energia potencial excedente DE é convertida em energia interna (Eint') e/ou energia translacional (Etr') dos produtos H+OH. Em todos os cálculos efectuados neste trabalho a molécula H2 foi preparada com energia vibracional Evib de 6.23 kcal mol-1 e energia rotacional Erot de 0.34 kcal mol-1 [o que corresponde à energia do estado quântico (v=0, j=1)], perfazendo um total de 6.57 kcal mol-1 para a energia interna Eint. A energia total do sistema (Etot) que se mantém constante ao longo de cada trajectória, depende então da energia de colisão, cujos valores variaram no intervalo 0.5 < Etr/kcal mol-1< 5. Em cada um dos casos foram calculadas 2000 trajectórias com um passo de integração Dt=0.1 u.a. e uma separação inicial O-H2 de r = 15 a0. Os resultados obtidos encontram-se sumariados na Tabela 2, sendo detalhadamente apresentados nas Figuras 6-10.
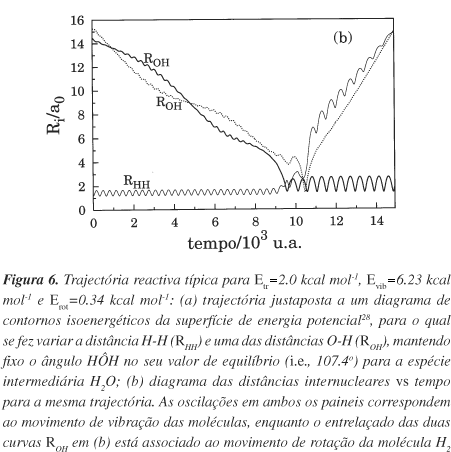
Na Figura 6 representa-se uma trajectória reactiva justaposta à superfície de energia potencial de Murrell e Carter28 [painel (a)] e o correspondente gráfico das distâncias internucleares vs tempo [painel (b)]. Deve notar-se que a trajectória escolhida conduz à formação de um complexo intermediário, correspondente ao poço de potencial [Figura 6(a)], que possui um tempo de vida de aproximadamente 1.5´103u.a.=0.04 ps [Figura 6(b)]. É ainda interessante notar que a amplitude (frequência) de vibração da espécie OH que se forma é muito maior (menor) que o valor correspondente para a molécula H2 dos reagentes (cƒ. as oscilações apresentadas pelas distâncias ROH e RHH na Figura 6), o que se deve ao facto da constante de força (massa reduzida) da primeira ser muito menor (maior) que a da segunda.
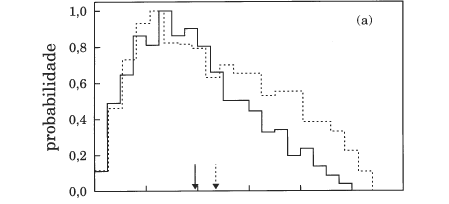
A função opacidade que se mostra na Figura 7 para Etr=0.5 e 5 kcal mol-1 apresenta um comportamento típico de uma reacção com um poço de potencial profundo (onde as forças de longo alcance desempenham um papel determinante)24 e em que, por isso, a etapa de captura do átomo de oxigénio é a dominante. Em ambos os casos, a probabilidade de reacção é próxima de 1 para todos os valores do parâmetro de impacto (b), caindo abruptamente para zero próximo de bmax. Uma vez que a reacção é dominada por forças de longo alcance, estas têm menos tempo para actuarem à medida que a energia de colisão aumenta e, portanto, o processo de captura torna-se menos eficaz para parâmetros de impacto grandes, fazendo diminuir o valor de bmax (ver também a Tabela 2).
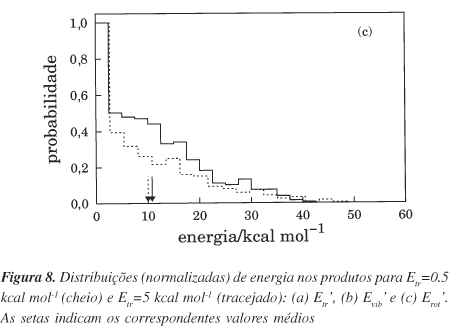
Na Figura 8 representam-se as distribuições das energias translacional [painel (a)], vibracional [painel (b)] e rotacional [painel (c)] nos produtos para as energias de colisão Etr=0.5 e 5 kcal mol-1; nesta figura mostram-se ainda os correspondentes valores médios. É interessante notar que o aumento da energia de colisão conduz à formação de produtos com um conteúdo energético na translação mais elevado, o que indica uma transferência de energia Etr®Etr' dos reagentes para os produtos. Também a energia vibracional média nos produtos é ligeiramente superior no caso Etr=5 kcal mol-1. De facto, esta energia de colisão origina uma distribuição [Figura 8(b)] onde são favorecidos valores de Evib' mais elevados, em comparação com Etr=0.5 kcal mol-1, o que indicia uma pequena transferência Etr®Evib' dos reagentes para os produtos. Contrariamente, o aumento de Etr leva a uma diminuição do número de trajectórias que formam produtos com energias rotacionais intermédias [Figura 8(c)], embora haja um ligeiro aumento de eventos reactivos que conduzem a valores de Erot' mais elevados. Em média, o resultado origina uma ligeira diminuição no valor médio de Erot', o que traduz uma transferência Etr®Erot' dos reagentes para os produtos praticamente inexistente.
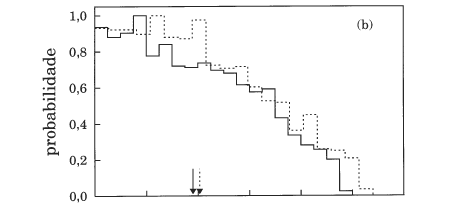
A secção eficaz diferencial constitui mais uma grandeza que pode elucidar sobre o mecanismo da reacção. A Figura 9 mostra as secções eficazes diferenciais para as energias de colisão Etr=0.5 e 5 kcal mol-1. Em ambos os casos, a secção eficaz diferencial apresenta dois picos, um para q'=0o e outro para q'=180o, correspondentes aos espalhamentos para a frente (forward) e para trás (backward), respectivamente. Isto significa que, uma vez formada a espécie intermediária H2O, é semelhante a probabilidade do produto OH se afastar de H no mesmo sentido ou em sentido contrário ao do átomo de oxigénio reagente que se aproxima de H2. Na realidade, a formação de um intermediário com um tempo de vida longo (regime osculatório) conduz a uma certa aleatoriedade na orientação do vector velocidade final.
Finalmente, na Figura 10 representam-se as secções eficazes para a reacção O(1D)+H2®OH+H; os valores numéricos encontram-se na Tabela 2. Pode notar-se nesta figura uma diminuição da secção eficaz de reacção com o aumento da energia de colisão, característico de processos cuja superfície de energia potencial não tem barreira ao longo do caminho de energia mínima e possui um poço associado a uma espécie intermediária (estável). De facto, como foi referido atrás, esta é uma reacção dominada pelo processo de captura do átomo de oxigénio, onde não é necessária energia translacional para vencer qualquer barreira. Assim, para energias de colisão baixas, as forças de longo alcance possibilitam aos reagentes reorientarem-se de acordo com o caminho mais favorável para que haja captura. À medida que a energia de colisão aumenta, o átomo de oxigénio aproxima-se mais rapidamente da molécula H2 (o tempo de colisão é encurtado), pelo que essas forças actuam durante menos tempo, podendo a aproximação dos reagentes ocorrer por regiões do potencial menos favoráveis, diminuindo então a probabilidade de reacção.
CONCLUSÕES
Apresentou-se uma descrição detalhada do método das trajectórias clássicas, para o que se considerou como ilustração uma reacção do tipo A+BC em 2D. A principal motivação foi a elaboração de um programa FORTRAN (traj2d.f) que se encontra disponível para a utilização por estudantes dos cursos de Química-Física avançada, Química Teórica e Química Computacional em www.qqesc.qui.uc.pt.
Escolheu-se como exemplo de aplicação a reacção O(1D)+H2® OH+H, a qual tem um grande interesse prático. Além de permitir ilustrar o uso da tecnologia envolvida num cálculo de trajectórias, serviu também para apresentar conceitos de grande importância em reactividade química.
AGRADECIMENTOS
Os autores agradecem o apoio financeiro da Fundação para a Ciência e Tecnologia, Portugal.
APÊNDICE
Álgebra envolvida na obtenção da Equação(36)
Considere-se a Equação (34). Diferenciando tal equação em ordem ao tempo, tem-se:

As relações entre
1 e P1, 1 e p1, e
1 e p1, e  1 e P1 são dadas pelas Equações (12), (13), e (14), respectivamente. De igual modo, podem obter-se relações idênticas entre 1´ e p1´, 1´ e P1´, e 1´ e P1´, diferenciando o Hamiltoniano da Equação (35) em ordem aos respectivos momentos:
1 e P1 são dadas pelas Equações (12), (13), e (14), respectivamente. De igual modo, podem obter-se relações idênticas entre 1´ e p1´, 1´ e P1´, e 1´ e P1´, diferenciando o Hamiltoniano da Equação (35) em ordem aos respectivos momentos:

Finalmente, a substituição adequada das Equações (12), (13), (14), (A.2), (A.3) e (A.4) na Equação (A.1) conduz à seguinte igualdade:

Resolvendo em ordem p1´, P1´ e P1´ o sistema de equações implícito na Equação (A.5), imediatamente se obtém a Equação (36).
Recebido em 13/9/02
Aceito em 10/12/02
- 1. Born, M.; Oppenheimer, J. R.; Ann. Phys. 1927, 85, 457.
- 2. Yarkony, D. R. Em Encyclopedia of Computational Chemistry; John Wiley & Sons, New York: 1998, p. 1894.
- 3. Varandas, A. J. C.; Marques, J. M. C.; J. Chem. Phys. 1994, 100, 1908.
- 4. Blais, N. C.; Zhao, M.; Mladenovic, M.; Truhlar, D. G.; Schwenke, D. W.; Sun, Y.; Kouri, D. J.; J. Chem. Phys. 1989, 91, 1038.
- 5. Karplus, M.; Porter, R. N.; Sharma, R.; J. Chem. Phys. 1965, 43, 3259.
- 6. Karplus, M.; Porter, R. N.; Sharma, R.; J. Chem. Phys. 1964, 40, 2033.
- 7. Karplus, M.; Raff, L. M.; J. Chem. Phys. 1964, 41, 1267.
- 8. Blais, N. C.; Bunker, D. L.; J. Chem. Phys. 1964, 41, 2377.
- 9. Bunker, D. L.; Meth. Comp. Physics 1971, 10, 287.
- 10. Muckerman, J. T.; J. Chem. Phys. 1971, 54, 1155.
- 11. Muckerman, J. T.; J. Chem. Phys. 1972, 56, 2997.
- 12. Raff, L. M.; Thompson, D. L.; Sims, L. B.; Porter, R. N.; J. Chem. Phys. 1972, 56, 5998.
- 13. Porter, R.; Raff, L. M. Em Dynamics of Molecular Collisions; Miller, W., ed.; Plenum: New York, 1976, p. 1.
- 14. Truhlar, D. G.; Muckerman, J. T. Em Atom-Molecule Collision Theory; Bernstein, R., ed.; Plenum: New York, 1979, p. 505.
- 15. Varandas, A. J. C.; Pais, A. A. C. C.; Marques, J. M. C.; Colóquio sobre Termodinâmica e Reactividade de Sistemas Moleculares, Memórias da Academia das Ciências de Lisboa, Academia das Ciências de Lisboa: Lisboa, 1994, p. 197.
- 16. Mayne, H. Em Dynamics of Molecules and Chemical Reactions; Wyatt, R. E.; Zhang, J. Z. H., eds.; Marcel Decker: New York, 1996, p. 589.
- 17. Varandas, A. J. C.; Int. Rev. Phys. Chem. 2000, 19, 199.
- 18. Reid, K. L.; Wheatley, R. J.; Horton, J. C.; Brydges, S. W.; J. Chem. Educ. 2000, 77, 407.
- 19. Hemphill, G. L.; White, J. M.; J. Chem. Educ. 1972, 49, 121.
- 20. Smith, I. W. M.; J. Chem. Educ. 1982, 59, 9.
- 21. Press, W. H.; Teukolski, S. A.; Vetterling, W. T.; Flannery, B. P.; Numerical Recipes in Fortran: the Art of Scientific Computing, Cambridge University Press: New York, 1992.
- 22. Raff, L. M.; Karplus, M.; J. Chem. Phys. 1966, 44, 1212.
- 23. Whitehead, J. C.; Mol. Phys. 1975, 29, 177.
- 24. Brouard, M.; Reaction Dynamics, Oxford University Press, 1998.
- 25. Eyring, H.; Lin, S. H.; Lin, S. M.; Basic Chemical Kinetics, John Wiley & Sons, 1980.
- 26. Schinke, R.; Lester, W. A.; J. Chem. Phys. 1980, 77, 3754.
- 27. Dunne, L. J.; Murrell, J. N.; Mol. Phys. 1983, 50, 635.
- 28. Murrell, J. N.; Carter, S.; Mills, I. M.; Guest, M. F.; Mol. Phys. 1981, 42, 605.
- 29. Murrell, J. N.; Carter, S.; J. Phys. Chem. 1984, 88, 4887.
- 30. Varandas, A. J. C.; J. Chem. Phys. 1996, 105, 3524.
- 31. Varandas, A. J. C.; J. Chem. Phys. 1997, 107, 867.
- 32. Murrell, J. N.; Carter, S.; Farantos, S. C.; Huxley, P.; Varandas, A. J. C.; Molecular potential energy functions, Wiley: Chichester, 1984.
- 33. Muckerman, J. T.; QCPE 1973, 11, 229.
- 34. Chapman, S.; Bunker, D. L.; Gelb, A.; QCPE 1975, 11, 273.
- 35. Hase, W. L.; QCPE, 453.
- 36. Hase, W. L.; Duchovic, R. J.; Hu, X.; Komornik, A.; Lim, K. F.; Lu, D.-H.; Peslherbe, G. H.; Swamy, K. N.; van de Linde, S. R.; Varandas, A. J. C.; Wang, H.; Wolf, R. J.; QCPE, 671.
Datas de Publicação
-
Publicação nesta coleção
14 Out 2003 -
Data do Fascículo
Out 2003
Histórico
-
Aceito
10 Dez 2002 -
Recebido
13 Set 2002