Abstracts
3b-hydroxysteroid dehydrogenase II (3β-HSD) deficiency represents a rare CAH variant. Newborns affected with its classic form have salt wasting in early infancy and genital ambiguity in both sexes. High levels of 17-hydroxypregnenolone (Δ517OHP) are characteristic, but extra-adrenal conversion to 17-hydroxyprogesterone (17OHP) may lead to positive results on newborn screening tests. Filter paper 17OHP on newborn screening test was performed by immunofluorometric assay, and serum determinations of 17OHP and Δ517OHP, by radioimmunoassay. A 46,XY infant with genital ambiguity and adrenal crisis at three months of age presented a positive result on newborn screening for CAH. Serum determinations of 17OHP and Δ517OHP were elevated, and a high Δ517OHP/cortisol relation was compatible with the diagnosis of 3β-HSD deficiency. Molecular analysis of the HSD3B2 gene from the affected case revealed the presence of the homozygous p.P222Q mutation, whereas his parents were heterozygous for it. We present the first report of 3β-HSD type II deficiency genotype-proven detected at the Newborn Screening Program in Brazil. The case described herein corroborates the strong genotype-phenotype correlation associated with the HSD3B2 p.P222Q mutation, which leads to a classic salt-wasting 3β-HSD deficiency. Further evaluation of 17OHP assays used in newborn screening tests would aid in determining their reproducibility, as well as the potential significance of moderately elevated 17OHP levels as an early indicator to the diagnosis of other forms of classic CAH, beyond 21-hydroxylase deficiency.
A deficiência da enzima 3β-hidroxiesteroide desidrogenase tipo 2 (3β-HSD) representa variante rara de hiperplasia adrenal congenital (HAC). Recém-nascidos afetados com a forma clássica apresentam perda de sal nas primeiras semanas de vida e ambiguidade genital em ambos os sexos. Concentrações elevadas de 17-hidroxipregnenolona (Δ517OHP) são características, porém sua conversão extra-adrenal a 17-hidroxiprogesterona (17OHP) pode resultar em resultados positivos no teste de triagem neonatal. A determinação da concentração de 17OHP obtida em amostra de sangue colhida em papel-filtro para triagem neonatal foi realizada por ensaio imunofluorimétrico, e as concentrações séricas de 17OHP and Δ517OHP, por radioimunoensaio. Um menino, 46,XY, com ambiguidade genital e crise adrenal aos 3 meses de vida, apresentou teste positivo na triagem neonatal para HAC. As concentrações séricas de 17OHP e Δ517OHP estavam aumentadas, bem como a relação Δ517OHP/cortisol, o que foi compatível com o diagnóstico de deficiência de 3β-HSD. A análise molecular do gene HSD3B2 revelou a mutação p.P222Q em homozigose na criança afetada e em heterozigose em seus pais, o que confirmou a deficiência de 3β-HSD com resultado moderadamente elevado na dosagem de 17OHP no “Teste do Pezinho” (Programa de Triagem Neonatal do Distrito Federal, Brasil). Esse caso corrobora a forte correlação genótipo-fenótipo associada à mutação p.P222Q no gene HSD3B2. Estudos futuros para avaliação dos ensaios utilizados na triagem neonatal para determinação de 17OHP poderão auxiliar na determinação do significado potencial de concentrações moderadamente elevadas de 17OHP como um indicador precoce para o diagnóstico de outras formas de HAC clássicas, além da deficiência de 21-hidroxilase.
INTRODUCTION
Congenital adrenal hyperplasia (CAH) is most commonly due to steroid-21-hydroxylase (CYP21) deficiency, which is considered one of the most common inborn errors of metabolism. The early recognition of its classical form is mandatory to prevent life-threatening salt-wasting adrenal crisis in the newborn. The diagnosis primarily relies on the finding of elevated plasmatic levels of 17OHprogesterone (17OHP), the immediate substrate of the enzyme CYP21 in the adrenal cortex. Neonatal screening has been widely shown to minimize delays in the diagnosis of CAH, thereby reducing morbidity and mortality related to the disease (11 . Speiser PW, White PC. Congenital adrenal hyperplasia. N Engl J Med. 2003;349:776-88.). Therefore, the determination of 17OHP level in dry blood spots collected in filter paper has been implemented in newborn screening programs worldwide.
Congenital adrenal hyperplasia has been included in the National Neonatal Screening Program
in Brazil in the year 2012, based on available epidemiological evidence about the relevance
and cost-effectiveness of the early case detection and treatment recommendations (22 . Portaria nº 2.829, December 14th, 2012. Ministry of Health. Available at:
bvsms.saude.gov.br/bvs/prt2829_14_12_2012.html. Accessed on: Oct 10,
2013.
bvsms.saude.gov.br/bvs/prt2829_14_12_201...
). Since then, many Neonatal Screening Reference Services
have been organized and started their work in different regions of the country, where
routine immunometric 17OHP determination has been offered for the entire population.
The estimated worldwide incidence of CAH due to CYP21 deficiency varies from 1:16.000 a 1:20.000 (33 . Krone N, Arlt W. Genetics of congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab. 2009;23:181-92.,44 . White PC. Neonatal screening for congenital adrenal hyperplasia. Nat Rev Endocrinol. 2009;5(9):490-8.). In a government-funded newborn screening program in the state of Goiás showed an incidence of 1:10,300 live births (55 . Silveira EL, dos Santos EP, Bachega TA, van der Linden Nader I, Gross JL, Elnecave RH. The actual incidence of congenital adrenal hyperplasia in Brazil may not be as high as inferred – an estimate based on a public neonatal screening program in the state of Goiás. J Pediatr Endocrinol Metab. 2008;21(5):455-60.). Less frequently, CAH may result from other enzymatic defects, leading to rare and variable clinical presentations, such as 11β-hydroxylase deficiency and, even more rarely, 3β-hydroxy-steroid-desidrogenase type II (3β-HSD) deficiency. Eventually, cases of less common forms of CAH have been described in which an elevated 17OHP level was firstly noted in a dry blood spot collected for neonatal screening for 21-hydroxylase deficiency (66 . Jeandron D, Sahakitrungruang T. A novel homozygous Q334X mutation in the HSD3B2 gene causing classic deficiency: an unexpected diagnosis after a positive newborn screen for 21-hydroxylase deficiency. Horm Res Paediatr. 2012;77:334-8.,77 . Nordenström A, Forest MG, Wedell A. A case of 3beta-hidroxysteroid dehydrogenase type II (HSD3B2) deficiency picked up by neonatal screening for 21-hydroxylase deficiency: difficulties and delay in etiologic diagnosis. Horm Res. 2007;68:204-8.).
3β-HSD deficiency results in salt-wasting or non-salt-wasting CAH with impaired
steroidogenesis in adrenals and gonads (88 . Simard J, Rheaume E, Sanchez R, Laflamme N, de Launoit Y, Luu-The V, et
al. Molecular basis of congenital adrenal hyperplasia due to 3beta-hydroxysteroid
dehydrogenase deficiency. Mol Endocrinol. 1993;7:716-28.,99 . Pang S. The molecular and clinical spectrum of 3beta-hydroxysteroid
dehydrogenase deficiency disorder. Trends Endocrinol Metab. 1998;9:82-6.). It also causes genital ambiguity, leading to slightly
virilized genitalia in genetic females and genital ambiguity in genetic males, explained by
the excess of weak adrenal androgens and gonadal testosterone deficiency, respectively
(99 . Pang S. The molecular and clinical spectrum of 3beta-hydroxysteroid
dehydrogenase deficiency disorder. Trends Endocrinol Metab. 1998;9:82-6.
10 . Pang S. Congenital adrenal hyperplasia owing to 3beta-hydroxysteroid
dehydrogenase deficiency. Endocrinol Metab Clin North Am. 2001;30:81-99.
11 . Mendonca B, Russel A, Vasconcelos-Leite M, Arnhold I, Bloise W,
Wajchenberg B, et al. Mutation in 3beta-hydroxysteroid dehydrogenase type II associated
with pseudohermaphroditism in males and premature pubarche or cryptic expression in
females. J Mol Endocrinol. 1994;12:119-22.
12 . Lutfallah C, Wang W, Mason JI, Chang YT, Haider A, Rich B, et al. Newly
proposed hormonal criteria via genotypic proof for type II 3beta-hydroxysteroid
dehydrogenase deficiency. J Clin Endocrinol Metab. 2002;87:2611-22.-1313 . Mermejo L, Elias L, Marui S, Moreira A, Mendonca B, de Castro M. Refining
hormonal diagnosis of type II 3beta-hydroxysteroid dehydrogenase deficiency in patients
with premature pubarche and hirsutism based on HSD3B2 genotyping. J Clin Endocrinol Metab.
2005;90(3):1287-93.).
Premature pubarche in young children and hirsutism and menstrual disorders during young
adulthood are believed to be equally present in non-salt-wasting forms of the disease, as
part of its clinical spectrum (1111 . Mendonca B, Russel A, Vasconcelos-Leite M, Arnhold I, Bloise W,
Wajchenberg B, et al. Mutation in 3beta-hydroxysteroid dehydrogenase type II associated
with pseudohermaphroditism in males and premature pubarche or cryptic expression in
females. J Mol Endocrinol. 1994;12:119-22.
12 . Lutfallah C, Wang W, Mason JI, Chang YT, Haider A, Rich B, et al. Newly
proposed hormonal criteria via genotypic proof for type II 3beta-hydroxysteroid
dehydrogenase deficiency. J Clin Endocrinol Metab. 2002;87:2611-22.-1313 . Mermejo L, Elias L, Marui S, Moreira A, Mendonca B, de Castro M. Refining
hormonal diagnosis of type II 3beta-hydroxysteroid dehydrogenase deficiency in patients
with premature pubarche and hirsutism based on HSD3B2 genotyping. J Clin Endocrinol Metab.
2005;90(3):1287-93.).
We present the first report of 3β-hydroxy-steroid dehydrogenase type II deficiency genotype-proven detected at the Newborn Screening Program in Brazil. The clinical and hormonal features of a 46, XY infant with a classical phenotype of 3β-HSD deficiency detected by a positive newborn screening test for CYP21 deficiency are described. Molecular analysis of the HSD3B2 gene revealed the homozygous P222Q mutation. This case highlights the significance of confirming the specific enzymatic deficiency with molecular genetic methods and the potential contribution of neonatal screening in predicting life-threatening salt-wasting complications, whatever the etiology of CAH.
CASE REPORT
The patient was an infant born at term after an uneventful gestation with genital ambiguity and palpable gonads, who had a positive neonatal screening result for CAH. At birth, weight was 3,970 g and height was 53 cm. Segmental physical examination was unremarkable except for a 2.5 cm phallus, with penoscrotal proximal hypospadia, incompletely fused labioscrotal folds and bilaterally palpable gonads. Karyotype was 46, XY. Neonatal 17OHP determination on a blood spot collected on filter paper S&S #903 (Guthrie test) on the first day of life was 5,170 ng/dL (normal value for weight at birth and age on the date of specimen collection: < 2,000 ng/dL) (14). There was no consanguinity or similar family history. Despite prior instructions and contact on the occasion of the screening result, the infant was brought to medical attention at three months of age with recurrent vomiting, dehydration, hyponatremia (Na = 107 mEq/L) and hypercalemia (K = 6,09 mEq/L). Serum 17OHP level was 2,430 ng/dL (< 200 ng/dL), whereas 17α-hydroxypregnenolone (Δ5 17OHP) was 1,080 ng/dL or 32.5 nmol/L (reference value < 10 ng/dL; proposed hormonal criteria for neonates with genotype-proven 3β-HSD deficiency: ≥ 84 nmol/L). Serum androstenedione was > 1,000 ng/dL (90-460 ng/dL), morning cortisol 5.29 µg/dL (0.145 µmol/L; reference value: 5-25 µg/dL) and ACTH 50.18 pg/mL (reference value: 0-46 pg/mL). The Δ5 17OHP/cortisol relation was 224, that is, above the proposed criteria for 3β-HSD deficiency diagnosis (≥ 94) (12). Hormonal levels were drawn just after starting vigorous fluid replacement and hydrocortisone and fludrocortisone supplementation, due to acute clinical instability. Subsequently, the patient fully recovered and has shown normal weight and height gain and psychomotor development suitable for age.
SUBJECTS AND METHODS
The study was approved by the Research Ethics Committee of the Faculty of Health Sciences, University of Brasilia, Brazil. Both parents gave informed consent for the participation of the patient in the study.
Hormonal determinations
Filter paper 17OHP determination was performed by an immunofluorometric assay (AutoDELFIA® Neonatal 17α-OH-progesterone), and serum determinations of 17OHP, Δ517OHP, androstenedione and cortisol, by radioimmunoassay.
Normal reference values were defined for each test by the manufacturer and validated by the reference laboratory.
Molecular genetics analysis
Genomic DNA from the index case was extracted from peripheral blood leucocytes by the salting-out method (1515 . Miller AS, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215.). The coding exons (exons 2 to 4) and exon-intron boundaries of the HSD3B2 gene were amplified using previously described primers (66 . Jeandron D, Sahakitrungruang T. A novel homozygous Q334X mutation in the HSD3B2 gene causing classic deficiency: an unexpected diagnosis after a positive newborn screen for 21-hydroxylase deficiency. Horm Res Paediatr. 2012;77:334-8.). PCR products were purified using the Qiagen purification kit (QIAquick®) and automatically sequenced in both sense and antisense orientations. Sequencing data were analysed using the software Sequencher version 5.0 (Gene Codes Corporation, Ann Arbor, Mich., USA) as compared with the HSD3B2 reference sequence (Gen Bank, ref. NG_013349.1).
Molecular results
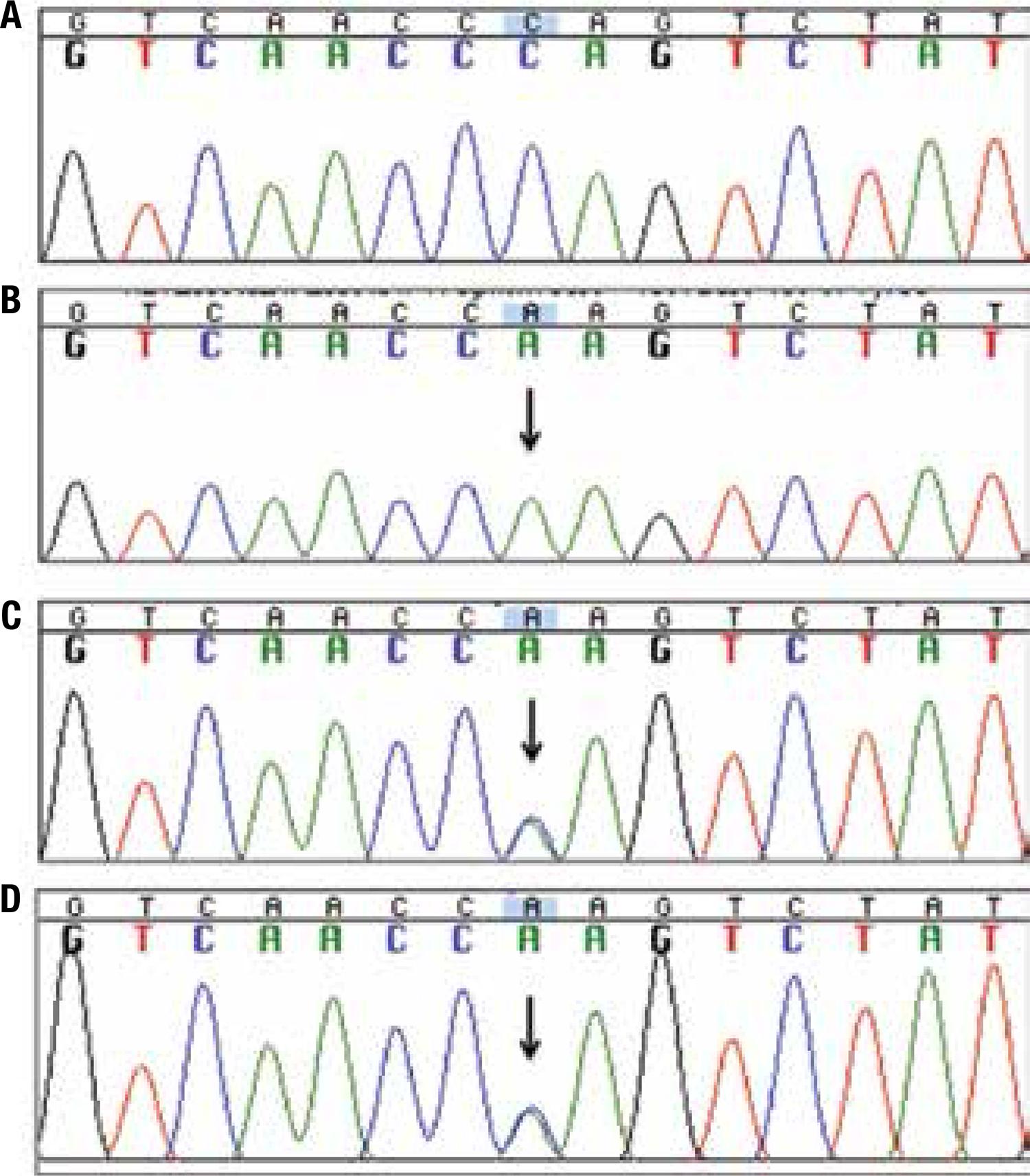
DNA sequencing of the HSD3B2 gene from the patient showed the homozygous change of a cytosine for an alanine at nucleotide 665, in exon 4. Both parents were heterozygous for the same missense mutation, c.665C>A, which results in the substitution of the aminoacid Proline to Glutamine at residue 222 (p.P222Q) (Figure 1).
Electropherogram of partial nucleotide sequence of the exon 4 of the HSD3B2 gene. (A) Wild-type control; (B) the patient, showing the homozygous mutation c.665C>A (p.P222Q) (arrow); (C) and (D) parents, heterozygotes.
DISCUSSION
We describe a Brazilian patient with a phenotype of classical 3β-HSD deficiency, who firstly presented with an abnormal 17OHP level on the newborn screening test for CAH due to steroid 21-hydroxylase deficiency. Molecular analysis of the HSD3B2 gene revealed that the patient was homozygous for the missense p.P222Q mutation, thereby confirming the clinical and hormonal diagnosis.
The 3β-hydroxysteroid dehydrogenase/Δ5-Δ4 isomerase (3β-HSD)
catalyzes the oxidation as well as the isomerization of 3β-hydroxy-5-ene steroids into
keto-4-ene steroids, allowing the synthesis of progesterone, 17α-hydroxyprogesterone,
androstenedione, and testosterone from their precursors pregnenolone,
17α-hydroxypregnenolone, dehydroepiandrosterone (DHEA), and 5-androstene-3β, 17β-diol
(Δ5-diol), respectively (1616 . Lachance Y, Luu-The V, Verrault H, Dumont M, LeBlanc G, Labrie F.
Structure of the human type II 3beta-hydroxysteroid dehydrogenase/Δ5-Δ4-isomerase (3βHSD)
gene: adrenal and gonadal specificity. DNA Cell Biol. 1991;10:701-11.,1717 . Luu-The V, Lachance Y, Labrie C, Leblanc G, Thomas JL, Strickler RC, et
al. Full-length cDNA structure and deduced amino acid sequence of human
3beta-hydroxy-5-ene steroid dehydrogenase. Mol Endocrinol. 1989;3:1310-2.). This reaction is a key step in the biosynthesis of
all classes of hormonal steroids: glucocorticoids, mineralocorticoids and sex steroids.
These hormones are essential for sexual differentiation, growth and development of many
human tissues. 3β-HSD expression is found in the placenta, adrenal cortex, ovary and testis,
but also in several peripheral tissues, including the skin, adipose tissue, breast, lung,
endometrium, prostate, liver, kidney, epididymis, and brain (33 . Krone N, Arlt W. Genetics of congenital adrenal hyperplasia. Best Pract
Res Clin Endocrinol Metab. 2009;23:181-92.,1616 . Lachance Y, Luu-The V, Verrault H, Dumont M, LeBlanc G, Labrie F.
Structure of the human type II 3beta-hydroxysteroid dehydrogenase/Δ5-Δ4-isomerase (3βHSD)
gene: adrenal and gonadal specificity. DNA Cell Biol. 1991;10:701-11.
17 . Luu-The V, Lachance Y, Labrie C, Leblanc G, Thomas JL, Strickler RC, et
al. Full-length cDNA structure and deduced amino acid sequence of human
3beta-hydroxy-5-ene steroid dehydrogenase. Mol Endocrinol. 1989;3:1310-2.
18 . Rheáume E, Lachance Y, Zhao H, Breton N, Dumont M, de Launoit Y, et al.
Structure and expression of a new DNA encoding the almost exclusive 3beta-hydroxysteroid
dehydrogenase/Δ5-Δ4-isomerase in human adrenals and gonads. Mol Endocrinol.
1991;5:1147-57.-1919 . Labrie F, Simard J, Luu-The V, Belanger A, Pelletier G. Structure,
function and tissue-specific gene expression of 3beta-hydroxysteroid dehydrogenase/D5-D4
isomerase enzymes in classical and peripheral intracrine steroidogenic tissues. J Steroid
Biochem Mol Biol. 1992;43:805-26.). Two isoforms of 3β-HSD have been described in humans (1717 . Luu-The V, Lachance Y, Labrie C, Leblanc G, Thomas JL, Strickler RC, et
al. Full-length cDNA structure and deduced amino acid sequence of human
3beta-hydroxy-5-ene steroid dehydrogenase. Mol Endocrinol. 1989;3:1310-2.,1818 . Rheáume E, Lachance Y, Zhao H, Breton N, Dumont M, de Launoit Y, et al.
Structure and expression of a new DNA encoding the almost exclusive 3beta-hydroxysteroid
dehydrogenase/Δ5-Δ4-isomerase in human adrenals and gonads. Mol Endocrinol.
1991;5:1147-57.). The type 1 enzyme is
expressed mainly in placenta and skin and is about five times more active than the type 2,
which is found in adrenals and gonads (1818 . Rheáume E, Lachance Y, Zhao H, Breton N, Dumont M, de Launoit Y, et al.
Structure and expression of a new DNA encoding the almost exclusive 3beta-hydroxysteroid
dehydrogenase/Δ5-Δ4-isomerase in human adrenals and gonads. Mol Endocrinol.
1991;5:1147-57.).
Paradoxically, in 3β-HSD deficiency increased circulating 17OHP levels are commonly found and are believed to result from the conversion of excessive amounts of accumulated Δ5 steroids (pregnenolone, 17OHpregnenolone and DHEA) by the isoenzyme 3β-hydroxysteroid-dehydrogenase type 1, in peripheral tissues. Similarly, circulating levels of Δ4 androstenedione and testosterone may be also increased.
In this context, although CAH screening programs are not aimed to detect different etiologies of CAH other than CYP21 deficiency, immunoassays used for 17OHP determination on newborn screening tests appear to allow the detection of some uncommon causes of CAH, such as 3β-HSD deficiency. However, the sensitivity of those assays for such less frequent enzymatic defects is still unclear (2020 . Kösel S, Burggraf S, Fingerhut R, Dörr HG, Roscher AA, Olgemöller B. Rapid second-tier molecular genetic analysis for congenital adrenal hyperplasia attributable to steroid 21-hydroxylase deficiency. Clin Chem. 2005;51:298-304.). Indeed, the main goal of CAH screening is the prompt detection of the classical form of CYP21 deficiency in order to prevent mortality due to severe salt-wasting crises in early life. False positive rates are still considerably high (11 . Speiser PW, White PC. Congenital adrenal hyperplasia. N Engl J Med. 2003;349:776-88.), despite great efforts to adjust cutoff values for some known interfering conditions. Nevertheless, the proportion of patients with other rare causes of CAH who would benefit from a positive screening test is still only presumptive. The adequate differentiation between these conditions and false positive results in infants with subtle clinical manifestations in the first weeks of life may be challenging and requires a high degree of suspicion by the care team.
Possibly, as far as screening programs become more widespread in the world, more such cases
will be identified. Accordingly, an increasing number of CAH cases caused by rare enzymatic
deficiencies have been described, and recently a significant number of them were picked up
by neonatal screening for 21-hydroxylase deficiency (66 . Jeandron D, Sahakitrungruang T. A novel homozygous Q334X mutation in the
HSD3B2 gene causing classic deficiency: an unexpected diagnosis after a positive newborn
screen for 21-hydroxylase deficiency. Horm Res Paediatr. 2012;77:334-8.,77 . Nordenström A, Forest MG, Wedell A. A case of 3beta-hidroxysteroid
dehydrogenase type II (HSD3B2) deficiency picked up by neonatal screening for
21-hydroxylase deficiency: difficulties and delay in etiologic diagnosis. Horm Res.
2007;68:204-8.,2020 . Kösel S, Burggraf S, Fingerhut R, Dörr HG, Roscher AA, Olgemöller B. Rapid
second-tier molecular genetic analysis for congenital adrenal hyperplasia attributable to
steroid 21-hydroxylase deficiency. Clin Chem. 2005;51:298-304.
21 . Jansen N, Riepe FG, Peter M, Sander S, Steuerwald U, Korsch E, et al.
Neonatal screening: identification of children with 11beta-hydroxylase deficiency by
second-tier testing. Horm Res Pediatr. 2012;77:195-9.-2222 . Johannsen TH, Mallet D, Dige-Petersen H, Múller J, Main KM, Morel Y, et
al. Delayed diagnosis of congenital adrenal hyperplasia with salt wasting due to type II
3beta-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab.
2005;90:2076-80.).
In the present case, the presence of genital ambiguity with palpable gonads and a moderately increased 17OHP on neonatal screening test immediately indicated the need to investigate 3β-HSD deficiency. Subsequently, the 46, XY karyotype, hormonal and molecular results confirmed the diagnosis. It is worth noting that, although clearly elevated, the measured serum concentration of Δ517OHPregnenolone (1,080 ng/dL or 32.5 nmol/L) was below the criteria proposed by Lutfallah and cols. (1212 . Lutfallah C, Wang W, Mason JI, Chang YT, Haider A, Rich B, et al. Newly proposed hormonal criteria via genotypic proof for type II 3beta-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab. 2002;87:2611-22.), reviewed in 2010: a baseline Δ5 17OHPreg ≥ 84 nmol/L for neonates, although the child was already 3 months old. A reason for this apparent discrepancy is related to the fact that the hormonal measurements were obtained just after the patient started intravenous hydrocortisone treatment during an acute adrenal crisis. Therefore, it is possible that higher Δ517OHPreg levels would have been noted earlier, if the tests were done before the acute crisis and treatment initiation. The clinical diagnosis of 3β-HSD deficiency in this case was then confirmed by the identification of the p.P222Q homozygous inactivating mutation in the HSD3B2 gene.
Several mutations, including frameshift, nonsense, in-frame deletions, splicing and
missense mutation, have been described in the HSD3B2 gene in patients
suffering from classical 3β-HSD deficiency (66 . Jeandron D, Sahakitrungruang T. A novel homozygous Q334X mutation in the
HSD3B2 gene causing classic deficiency: an unexpected diagnosis after a positive newborn
screen for 21-hydroxylase deficiency. Horm Res Paediatr. 2012;77:334-8.,2323 . Simard J, Ricketts ML, Gingras S, Soucy P, Feltus FA, Melner MH. Molecular
biology of the 3beta-hydroxysteroid dehydrogenase/Δ5-Δ4-isomerase Gene Family. Endocr Rev.
2005;26(4):525-82.
24 . Rheaume E, Simard J, Morel Y, Mebarki F, Zachmann M, Forest MG, et al.
Congenital adrenal hyperplasia due to point mutations in the type II 3beta-hydroxysteroid
dehydrogenase gene. Nat Genet. 1992;1:239-45.-2525 . Moisan AM, Ricketts ML, Tardy V, Desrochers M, Mebarki F, Chaussain JL, et
al. New insight into the molecular basis of 3beta-hydroxysteroid dehydrogenase deficiency:
identification of eight mutations in the HSD3B2 gene in eleven patients from seven new
families and comparison of the functional properties of twenty-five mutant enzymes. J Clin
Endocrinol Metab. 1999;84:4410-25.). The
functional characterization of these mutant proteins has helped understanding the
structure-function relationships of the 3β-HSD family (2121 . Jansen N, Riepe FG, Peter M, Sander S, Steuerwald U, Korsch E, et al.
Neonatal screening: identification of children with 11beta-hydroxylase deficiency by
second-tier testing. Horm Res Pediatr. 2012;77:195-9.,2424 . Rheaume E, Simard J, Morel Y, Mebarki F, Zachmann M, Forest MG, et al.
Congenital adrenal hyperplasia due to point mutations in the type II 3beta-hydroxysteroid
dehydrogenase gene. Nat Genet. 1992;1:239-45.
25 . Moisan AM, Ricketts ML, Tardy V, Desrochers M, Mebarki F, Chaussain JL, et
al. New insight into the molecular basis of 3beta-hydroxysteroid dehydrogenase deficiency:
identification of eight mutations in the HSD3B2 gene in eleven patients from seven new
families and comparison of the functional properties of twenty-five mutant enzymes. J Clin
Endocrinol Metab. 1999;84:4410-25.
26 . Lusa ALG, Lemos-Marini SHV, Soardi FC, Ferraz LFC, Guerra-Júnior G, Mello
MP. Structural aspects of the p.P222Q homozygous mutation of HSD3B2 gene in a patient with
congenital adrenal hyperplasia. Arq Bras Endocrinol Metab.
2010;54(8):768-74.-2727 . Thomas J, Duax W, Addlagatta A, Kacsoh B, Brandt S, Norris W.
Structure/function aspects of human 3beta-hydroxysteroid dehydrogenase. Mol Cell
Endocrinol. 2004;215:73-82.). The p.P222Q mutation found in the present case leads to a shift in amino acid
interactions and change the substrate’s binding site, thereby significantly reducing protein
activity (2626 . Lusa ALG, Lemos-Marini SHV, Soardi FC, Ferraz LFC, Guerra-Júnior G, Mello
MP. Structural aspects of the p.P222Q homozygous mutation of HSD3B2 gene in a patient with
congenital adrenal hyperplasia. Arq Bras Endocrinol Metab.
2010;54(8):768-74.). This variant affects a highly
conserved amino acid in mammals, a fact that reinforces its importance for the function of
the enzyme (2626 . Lusa ALG, Lemos-Marini SHV, Soardi FC, Ferraz LFC, Guerra-Júnior G, Mello
MP. Structural aspects of the p.P222Q homozygous mutation of HSD3B2 gene in a patient with
congenital adrenal hyperplasia. Arq Bras Endocrinol Metab.
2010;54(8):768-74.,2828 . McBride M, McVie A, Burridge S, Brintnell B, Craig N, Wallace A, et al.
Cloning, expression, and physical mapping of the 3beta-hydroxysteroid dehydrogenase gene
cluster (HSD3BP1-HSD3BP5) in human. Genomics. 1999;61:277-84.
29 . Welzel M, Wüstemann N, Simic-Schleicher G, Dörr HG, Schulze E, Shaikh G,
et al. Carboxyl-terminal mutations in 3beta-hydroxysteroid dehydrogenase type II cause
severe salt-wasting congenital adrenal hyperplasia. J Clin Endocrinol Metab.
2008;93:1418-25.-3030 . Morel Y, Mébarki F, Rhéaume E, Sanchez R, Forest M, Simard J.
Structure-function relationships of 3beta-hydroxysteroid dehydrogenase deficiency.
Steroids. 1997;62:176-84.).
The p.P222Q mutation in the HSD3B2 gene has been firstly described in an Argelian family, in which a 46,XY boy and his 46,XX sister presented with a classical phenotype of 3β-HSD deficiency (2525 . Moisan AM, Ricketts ML, Tardy V, Desrochers M, Mebarki F, Chaussain JL, et al. New insight into the molecular basis of 3beta-hydroxysteroid dehydrogenase deficiency: identification of eight mutations in the HSD3B2 gene in eleven patients from seven new families and comparison of the functional properties of twenty-five mutant enzymes. J Clin Endocrinol Metab. 1999;84:4410-25.). Since then, it has been reported only in other two cases from Brazil, with similar clinical presentations (1111 . Mendonca B, Russel A, Vasconcelos-Leite M, Arnhold I, Bloise W, Wajchenberg B, et al. Mutation in 3beta-hydroxysteroid dehydrogenase type II associated with pseudohermaphroditism in males and premature pubarche or cryptic expression in females. J Mol Endocrinol. 1994;12:119-22.,1313 . Mermejo L, Elias L, Marui S, Moreira A, Mendonca B, de Castro M. Refining hormonal diagnosis of type II 3beta-hydroxysteroid dehydrogenase deficiency in patients with premature pubarche and hirsutism based on HSD3B2 genotyping. J Clin Endocrinol Metab. 2005;90(3):1287-93.,2626 . Lusa ALG, Lemos-Marini SHV, Soardi FC, Ferraz LFC, Guerra-Júnior G, Mello MP. Structural aspects of the p.P222Q homozygous mutation of HSD3B2 gene in a patient with congenital adrenal hyperplasia. Arq Bras Endocrinol Metab. 2010;54(8):768-74.,3131 . Marui S, Castro M, Latronico AC, Elias LLK, Arnhold IJP, Moreira AC, et al. Mutations in the type II 3beta-hydroxysteroid dehydrogenase (HSD3B2) gene can cause premature pubarche in girls. Clin Endocrinol. 2000;52:67-75.). A different mutation was also described in the same position at the protein, the p.P222T, and was associated with premature pubarche, but not salt-wasting form. Notably, the mutated protein 222T was not detected on in vitro assays with transiently transfected 293 cells, bringing up an additional pathogenic mechanism of a severe impairment in protein stability (3232 . Pang S, Wang W, Rich B, David R, Chang YT, Carbunaru G, et al. A novel nonstop mutation in the stop codon and a novel missense mutation in the type II 3beta-hydroxysteroid dehydrogenase (3beta-HSD) gene causing, respectively, nonclassic and classic 3beta-HSD deficiency congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2002;87(6):2556-63.).
In summary, the case described herein corroborates the strong genotype-phenotype correlation associated with the p.P222Q mutation in the HSD3B2 gene, which leads to a classical salt-wasting 3β-HSD deficiency. The finding of a positive result on neonatal screening test for CAH in a 46, XY infant with ambiguous genitalia strongly suggests the diagnosis of 3β-HSD deficiency and may prevent morbity and mortality associated with the occurrence of a salt-loosing crisis, if promptly recognized. Further evaluation of 17OHP assays used in newborn screening tests would aid in determining their reproducibility, as well as the potential significance of moderately elevated 17OHP levels as an early indicator to the diagnosis of other forms of classic CAH, beyond 21-hydroxylase deficiency.
REFERENCES
-
1Speiser PW, White PC. Congenital adrenal hyperplasia. N Engl J Med. 2003;349:776-88.
-
2Portaria nº 2.829, December 14th, 2012. Ministry of Health. Available at: bvsms.saude.gov.br/bvs/prt2829_14_12_2012.html. Accessed on: Oct 10, 2013.
» bvsms.saude.gov.br/bvs/prt2829_14_12_2012.html -
3Krone N, Arlt W. Genetics of congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab. 2009;23:181-92.
-
4White PC. Neonatal screening for congenital adrenal hyperplasia. Nat Rev Endocrinol. 2009;5(9):490-8.
-
5Silveira EL, dos Santos EP, Bachega TA, van der Linden Nader I, Gross JL, Elnecave RH. The actual incidence of congenital adrenal hyperplasia in Brazil may not be as high as inferred – an estimate based on a public neonatal screening program in the state of Goiás. J Pediatr Endocrinol Metab. 2008;21(5):455-60.
-
6Jeandron D, Sahakitrungruang T. A novel homozygous Q334X mutation in the HSD3B2 gene causing classic deficiency: an unexpected diagnosis after a positive newborn screen for 21-hydroxylase deficiency. Horm Res Paediatr. 2012;77:334-8.
-
7Nordenström A, Forest MG, Wedell A. A case of 3beta-hidroxysteroid dehydrogenase type II (HSD3B2) deficiency picked up by neonatal screening for 21-hydroxylase deficiency: difficulties and delay in etiologic diagnosis. Horm Res. 2007;68:204-8.
-
8Simard J, Rheaume E, Sanchez R, Laflamme N, de Launoit Y, Luu-The V, et al. Molecular basis of congenital adrenal hyperplasia due to 3beta-hydroxysteroid dehydrogenase deficiency. Mol Endocrinol. 1993;7:716-28.
-
9Pang S. The molecular and clinical spectrum of 3beta-hydroxysteroid dehydrogenase deficiency disorder. Trends Endocrinol Metab. 1998;9:82-6.
-
10Pang S. Congenital adrenal hyperplasia owing to 3beta-hydroxysteroid dehydrogenase deficiency. Endocrinol Metab Clin North Am. 2001;30:81-99.
-
11Mendonca B, Russel A, Vasconcelos-Leite M, Arnhold I, Bloise W, Wajchenberg B, et al. Mutation in 3beta-hydroxysteroid dehydrogenase type II associated with pseudohermaphroditism in males and premature pubarche or cryptic expression in females. J Mol Endocrinol. 1994;12:119-22.
-
12Lutfallah C, Wang W, Mason JI, Chang YT, Haider A, Rich B, et al. Newly proposed hormonal criteria via genotypic proof for type II 3beta-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab. 2002;87:2611-22.
-
13Mermejo L, Elias L, Marui S, Moreira A, Mendonca B, de Castro M. Refining hormonal diagnosis of type II 3beta-hydroxysteroid dehydrogenase deficiency in patients with premature pubarche and hirsutism based on HSD3B2 genotyping. J Clin Endocrinol Metab. 2005;90(3):1287-93.
-
14Olgemöller B, Roscher AA, Liebl B, Fingerhut R. Screening for congenital adrenal hyperplasia: adjustment for 17-hydroxyprogesterone cut-off values to both age and birth weight markedly improves the predictive value. J Clin Endocrinol Metab. 2003;88(12):5790-94.
-
15Miller AS, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215.
-
16Lachance Y, Luu-The V, Verrault H, Dumont M, LeBlanc G, Labrie F. Structure of the human type II 3beta-hydroxysteroid dehydrogenase/Δ5-Δ4-isomerase (3βHSD) gene: adrenal and gonadal specificity. DNA Cell Biol. 1991;10:701-11.
-
17Luu-The V, Lachance Y, Labrie C, Leblanc G, Thomas JL, Strickler RC, et al. Full-length cDNA structure and deduced amino acid sequence of human 3beta-hydroxy-5-ene steroid dehydrogenase. Mol Endocrinol. 1989;3:1310-2.
-
18Rheáume E, Lachance Y, Zhao H, Breton N, Dumont M, de Launoit Y, et al. Structure and expression of a new DNA encoding the almost exclusive 3beta-hydroxysteroid dehydrogenase/Δ5-Δ4-isomerase in human adrenals and gonads. Mol Endocrinol. 1991;5:1147-57.
-
19Labrie F, Simard J, Luu-The V, Belanger A, Pelletier G. Structure, function and tissue-specific gene expression of 3beta-hydroxysteroid dehydrogenase/D5-D4 isomerase enzymes in classical and peripheral intracrine steroidogenic tissues. J Steroid Biochem Mol Biol. 1992;43:805-26.
-
20Kösel S, Burggraf S, Fingerhut R, Dörr HG, Roscher AA, Olgemöller B. Rapid second-tier molecular genetic analysis for congenital adrenal hyperplasia attributable to steroid 21-hydroxylase deficiency. Clin Chem. 2005;51:298-304.
-
21Jansen N, Riepe FG, Peter M, Sander S, Steuerwald U, Korsch E, et al. Neonatal screening: identification of children with 11beta-hydroxylase deficiency by second-tier testing. Horm Res Pediatr. 2012;77:195-9.
-
22Johannsen TH, Mallet D, Dige-Petersen H, Múller J, Main KM, Morel Y, et al. Delayed diagnosis of congenital adrenal hyperplasia with salt wasting due to type II 3beta-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab. 2005;90:2076-80.
-
23Simard J, Ricketts ML, Gingras S, Soucy P, Feltus FA, Melner MH. Molecular biology of the 3beta-hydroxysteroid dehydrogenase/Δ5-Δ4-isomerase Gene Family. Endocr Rev. 2005;26(4):525-82.
-
24Rheaume E, Simard J, Morel Y, Mebarki F, Zachmann M, Forest MG, et al. Congenital adrenal hyperplasia due to point mutations in the type II 3beta-hydroxysteroid dehydrogenase gene. Nat Genet. 1992;1:239-45.
-
25Moisan AM, Ricketts ML, Tardy V, Desrochers M, Mebarki F, Chaussain JL, et al. New insight into the molecular basis of 3beta-hydroxysteroid dehydrogenase deficiency: identification of eight mutations in the HSD3B2 gene in eleven patients from seven new families and comparison of the functional properties of twenty-five mutant enzymes. J Clin Endocrinol Metab. 1999;84:4410-25.
-
26Lusa ALG, Lemos-Marini SHV, Soardi FC, Ferraz LFC, Guerra-Júnior G, Mello MP. Structural aspects of the p.P222Q homozygous mutation of HSD3B2 gene in a patient with congenital adrenal hyperplasia. Arq Bras Endocrinol Metab. 2010;54(8):768-74.
-
27Thomas J, Duax W, Addlagatta A, Kacsoh B, Brandt S, Norris W. Structure/function aspects of human 3beta-hydroxysteroid dehydrogenase. Mol Cell Endocrinol. 2004;215:73-82.
-
28McBride M, McVie A, Burridge S, Brintnell B, Craig N, Wallace A, et al. Cloning, expression, and physical mapping of the 3beta-hydroxysteroid dehydrogenase gene cluster (HSD3BP1-HSD3BP5) in human. Genomics. 1999;61:277-84.
-
29Welzel M, Wüstemann N, Simic-Schleicher G, Dörr HG, Schulze E, Shaikh G, et al. Carboxyl-terminal mutations in 3beta-hydroxysteroid dehydrogenase type II cause severe salt-wasting congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2008;93:1418-25.
-
30Morel Y, Mébarki F, Rhéaume E, Sanchez R, Forest M, Simard J. Structure-function relationships of 3beta-hydroxysteroid dehydrogenase deficiency. Steroids. 1997;62:176-84.
-
31Marui S, Castro M, Latronico AC, Elias LLK, Arnhold IJP, Moreira AC, et al. Mutations in the type II 3beta-hydroxysteroid dehydrogenase (HSD3B2) gene can cause premature pubarche in girls. Clin Endocrinol. 2000;52:67-75.
-
32Pang S, Wang W, Rich B, David R, Chang YT, Carbunaru G, et al. A novel nonstop mutation in the stop codon and a novel missense mutation in the type II 3beta-hydroxysteroid dehydrogenase (3beta-HSD) gene causing, respectively, nonclassic and classic 3beta-HSD deficiency congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2002;87(6):2556-63.
Publication Dates
-
Publication in this collection
Aug 2014
History
-
Received
27 Oct 2013 -
Accepted
24 Mar 2014