
Abstract
The immunomodulador glatiramer acetate (GA) has been shown to significantly reduce the severity of symptoms during the course of multiple sclerosis and in its animal model - experimental autoimmune encephalomyelitis (EAE). Since GA may influence the response of non-neuronal cells in the spinal cord, it is possible that, to some extent, this drug affects the synaptic changes induced during the exacerbation of EAE. In the present study, we investigated whether GA has a positive influence on the loss of inputs to the motoneurons during the course of EAE in rats. Lewis rats were subjected to EAE associated with GA or placebo treatment. The animals were sacrificed after 15 days of treatment and the spinal cords processed for immunohistochemical analysis and transmission electron microscopy. A correlation between the synaptic changes and glial activation was obtained by performing labeling of synaptophysin and glial fibrillary acidic protein using immunohistochemical analysis. Ultrastructural analysis of the terminals apposed to alpha motoneurons was also performed by electron transmission microscopy. Interestingly, although the GA treatment preserved synaptophysin labeling, it did not significantly reduce the glial reaction, indicating that inflammatory activity was still present. Also, ultrastructural analysis showed that GA treatment significantly prevented retraction of both F and S type terminals compared to placebo. The present results indicate that the immunomodulator GA has an influence on the stability of nerve terminals in the spinal cord, which in turn may contribute to its neuroprotective effects during the course of multiple sclerosis.
Experimental autoimmune encephalomyelitis; Spinal cord; Synapse elimination; Motoneuron; Immunomodulator; Glatiramer acetate
Abstract
Introduction
Material and Methods
Results
Discussion
Braz J Med Biol Res, February 2009, Volume 42(2) 179-188
The immunomodulator glatiramer acetate influences spinal motoneuron plasticity during the course of multiple sclerosis in an animal model
K.B. Marques1, J.M. Scorisa1, R. Zanon1, C.M. Freria1, L.M.B. Santos2, B.P. Damasceno3 and A.L.R. Oliveira1
1Departamento de Anatomia, 2Departamento de Microbiologia e Imunologia, Instituto de Biologia, 3Departamento de Neurologia, Instituto de Ciências Médicas, Universidade Estadual de Campinas, Campinas, SP, Brasil
 References
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
References
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
The immunomodulador glatiramer acetate (GA) has been shown to significantly reduce the severity of symptoms during the course of multiple sclerosis and in its animal model - experimental autoimmune encephalomyelitis (EAE). Since GA may influence the response of non-neuronal cells in the spinal cord, it is possible that, to some extent, this drug affects the synaptic changes induced during the exacerbation of EAE. In the present study, we investigated whether GA has a positive influence on the loss of inputs to the motoneurons during the course of EAE in rats. Lewis rats were subjected to EAE associated with GA or placebo treatment. The animals were sacrificed after 15 days of treatment and the spinal cords processed for immunohistochemical analysis and transmission electron microscopy. A correlation between the synaptic changes and glial activation was obtained by performing labeling of synaptophysin and glial fibrillary acidic protein using immunohistochemical analysis. Ultrastructural analysis of the terminals apposed to alpha motoneurons was also performed by electron transmission microscopy. Interestingly, although the GA treatment preserved synaptophysin labeling, it did not significantly reduce the glial reaction, indicating that inflammatory activity was still present. Also, ultrastructural analysis showed that GA treatment significantly prevented retraction of both F and S type terminals compared to placebo. The present results indicate that the immunomodulator GA has an influence on the stability of nerve terminals in the spinal cord, which in turn may contribute to its neuroprotective effects during the course of multiple sclerosis.
Key words: Experimental autoimmune encephalomyelitis; Spinal cord; Synapse elimination; Motoneuron; Immunomodulator; Glatiramer acetate
Multiple sclerosis (MS) is a central nervous system demyelinating and degenerative disease of unknown etiology, although, once initiated, it actively involves the immune system. Demyelination similar to that seen during the course of MS may be induced in animals, in a model called experimental autoimmune encephalomyelitis (EAE) (1-5). During the course of EAE, damage to the axons as well as the occurrence of synaptic plasticity phenomena in the brain and spinal cord contribute to the clinical signs of the disease (3,6,7). Synaptic plasticity in the spinal cord may explain, at least in part, the rapid sensorimotor recovery of the animals soon after tetraplegia (3,5).
In view of the importance of a better understanding of MS as well as its experimental models, major advances in understanding how cellular and humoral immune responses contribute to the pathogenesis of these diseases have been made in recent years (8-12). Thus, targeting key elements of the immunological cascade that culminate in neural and glial tissue damage may offer a number of advantages over currently available treatment strategies (13-17).
Glatiramer acetate (GA), an approved drug for the treatment of MS, displays a variety of immunomodulatory effects, decreasing the intensity and frequency of the episodes (18,19). Although its mechanism of action is not completely known, its inhibition of central nervous system myelin antigens by competing as an MHC ligand may be of importance (20-22). The ability to stimulate proliferation of Th2 over Th1 cells, suppressing T-cell activity is also of relevance as well (23). Since GA may influence the response of non-neuronal cells in the spinal cord, this drug may affect, to some extent, the synaptic changes induced during the exacerbation of EAE. This hypothesis has been explored in just a few studies dealing with optic nerve injury or glutamate toxicity and in motoneuron disease models (24-26).
Nevertheless, the impact of GA on nervous tissue, especially on neurons during the course of the disease, is virtually unknown. The fact that GA influences cell proliferation as well as neurotrophic factor synthesis (27) indicates that the drug may have a role in synaptic elimination/retraction, changing and reducing the seriousness of the course of the disease.
Taking into account the possible influence of GA on plastic events in the spinal cord during the course of EAE, the present study investigated the synaptic elimination process as well as the astroglial reaction in the spinal cord during the different stages of the disease, using treatment on a daily basis with the drug.
Animals
Forty adult female Lewis rats, 7 weeks old (~250 g body weight), were obtained from the Multidisciplinary Center for Biological Investigation (CEMIB/Unicamp) and housed on a 12-h light/dark cycle with free access to food and water. The Institutional Committee for Ethics in Animal Experimentation (CEEA/IB/Unicamp, Proc. #794-2) approved the study, and the experiments were performed in accordance with the guidelines of the Brazilian College for Animal Experimentation (COBEA - http://www.cobea. org.br/index.php?pg=Principios%20Eticos).
Induction of EAE
The rats were immunized with a single injection of 100 µL guinea pig myelin basic protein (25 µg, Sigma, USA) associated with heat-inactivated Mycobacterium tuberculosis H37RA (2.0 mg/mL; Difco Laboratories, USA) and complete Freund's adjuvant emulsion. This solution was injected subcutaneously into the footpad of a hind leg. A daily analysis and scoring of the signs and symptoms of EAE severity was carried out as follows: grade 0, no clinical signs; grade 1, tail weakness or paralysis; grade 2, hind leg paraparesis; grade 3, hind leg paralysis; grade 4, complete paralysis (tetraplegia). In rats, EAE develops as an acute monophasic disease, so that after the peak (or exacerbation) it regresses spontaneously within 5 days. The entire course of the disease from immunization to the initial recovery from paralysis (absence of clinical signs) takes about 14 days. In the present study, the analyses were performed when the EAE-placebo group reached the peak of the disease. At that point, all animals of the three experimental groups were sacrificed. However, in order to have a complete course of the disease with and without the treatment with GA, two extra groups of EAE-placebo and EAE-GA were followed up to the 30th day after immunization (Figure 3I and J).
Glatiramer acetate treatment
GA (batch 334218; Teva Pharmaceutical Industries, Israel) was used throughout the study. GA treatment was performed daily by subcutaneous injection of Copaxone (0.5 mg·animal-1·day-1) in sodium phosphate buffer (100 µL), pH 7.4, for 2 weeks. GA treatment (N = 10) was started 30 min after the procedures for EAE induction, being repeated daily by subcutaneous injections for two weeks. The controls were similarly treated with placebo (buffered saline, 100 µL).
Specimen preparation
Two weeks after the induction of EAE, the animals were anesthetized with a mixture of 10 mg/kg kensol (xylazine, Köning, Argentina) and 50 mg/kg vetaset (Cetamine, USA) and the vascular system was rinsed by transcardial perfusion with sodium phosphate buffer, pH 7.4. For the immunohistochemical detection of synaptophysin and glial fibrillary acidic protein (GFAP), the rats were fixed by vascular perfusion with 4% paraformaldehyde in sodium phosphate buffer, pH 7.4. Thereafter, the lumbar intumescence was dissected out, postfixed overnight and then frozen. For electron microscopy, 100 mL of a fixative containing 2.5% glutaraldehyde and 0.5% paraformaldehyde in sodium phosphate buffer, pH 7.4, were perfused through the ascending aorta as previously described (5). The lumbar spinal cord was removed and stored overnight in the same fixative at 4°C. The specimens were then trimmed, post-fixed with osmium tetroxide, dehydrated and embedded in Durcupan (Fluka, Switzerland). Ultra-thin sections from the L4-L6 segments were collected on formvar-coated copper grids, contrasted with uranyl acetate and lead citrate, and examined under a Leo906 transmission electron microscope operated at 60 KV.
Immunohistochemistry
Transverse sections of the spinal cord (12 µm-thick) were incubated overnight in a moist chamber at 4°C with the following primary antibodies: human anti-synaptophysin (Dako, Denmark, 1:100 in 3% BSA and 0.2% Triton-X in 10 mM sodium phosphate buffer, pH 7.4) was used to investigate the synaptic covering, since it is a synaptic glycoprotein found in the vesicle membrane of pre-synaptic terminals. Goat anti-GFAP (Santa Cruz, USA, 1:200 in 3% BSA and 0.2% Triton-X in 10 mM sodium phosphate buffer, pH 7.4) was used to analyze astrocyte reactivity. GFAP is an intermediate filament that is up-regulated during astrogliosis. After rinsing, the secondary antibodies were applied and incubated for 45 min, according to the primary antibody hosts (CY-2 and CY-3, Jackson ImmunoResearch, USA; 1:250). The sections were then rinsed in sodium phosphate buffer, pH 7.4, mounted in a mixture of glycerol/phosphate buffer (3:1) and observed with a laser scanning confocal microscope (Bio-Rad, USA), mounted on an Axiovert 135 inverted microscope (Zeiss, Germany). The excitation wavelength was 488 nm for CY-2 fluorescence and 568 nm for CY-3 fluorescence. For quantitative measurements (synaptophysin and GFAP immunoreactivity), three representative images of the ventral horn were captured at a final magnification of 400X, with the microscope settings being kept the same for all slides. A total of 15 sampled images from each specimen per group were acquired and the quantification was performed with the enhanced contrast and density slicing feature of the IMAGEJ software (version 1.33u, National Institutes of Health, USA). The integrated density of pixels was systematically measured in six representative areas of the lateral motor nucleus from each section, as previously described (28). The mean and standard error were calculated for each group and statistical analysis was performed.
Analysis of inflammation and demyelination during the course of EAE
The Sudan black technique was used to demonstrate the demyelination process during disease development. For this purpose, frozen sections were kept at room temperature, rinsed with 10 mM sodium phosphate buffer, pH 7.4, and stained with Sudan black in 70% ethanol for 30 min. The specimens were differentiated in 50% ethanol, rinsed with distilled water and mounted in a mixture of glycerol/phosphate buffer (3:1).
The influx of lymphocytes close to the spinal motoneurons was studied on semi-thin Toluidine blue-stained sections obtained prior to the ultra-thin sections.
Analysis of the ultra-thin sections
Neurons with large cell bodies (average of 35 mm in diameter) found in the sciatic motoneuron pool and cut along the nuclear plane were identified as alpha motoneurons based on the presence of C-type nerve terminals. The surface of the cells was then sequentially digitalized at a magnification of 10,000 using a video camera connected to a computerized system and the acquisition feature of the Kontron KS300 software (Zeiss). The images were then mounted together using a vectorial software. Synaptic terminals apposing the motoneuron somata were identified and their numbers per 100 mm of cell membrane and the membrane covering of all terminals in percent of membrane length were calculated using the measurement tool of the Image Tool software (Version 3.0, The University of Texas Health Center in Santo Antonio, USA). The terminals were classified as type F (with flattened synaptic vesicles, inhibitory synapses containing glycine and GABA), type S (with spherical synaptic vesicles, excitatory synapses containing glutamate) and type C (with a subsynaptic cistern, cholinergic synapse), according to the nomenclature by Conradi and Skoglund (29). The distance between consecutive nerve terminals covering the motoneurons was also determined. A total of 40 sciatic motoneurons (2 neurons/animal in four groups of 5 animals: normal; EAE-induced, grade 3/exacerbation; EAE-induced and GA-treated; EAE-induced and placebo-treated) were studied. Only large neurons present in the sciatic motor pool and presenting at least one C-terminal were selected for the ultrastructural analysis.
Statistical analysis
Data were analyzed by ANOVA and the results were compared among groups by the two-tailed Student t-test for parametric data or by the two-tailed Mann-Whitney U-test for non-parametric data at P < 0.05 and P < 0.01. All data are reported as mean ± SEM.
Preserved synaptophysin immunolabeling and decreased glial reaction after GA treatment
Figure 1 shows synaptophysin and GFAP immunostaining. Quantitative measurements were made in the spinal cord motor nuclei of all experimental groups and are presented in Figure 2. The basal immunolabeling was obtained from normal specimens showing relatively intense synaptophysin staining (11.54 ± 0.98; mean integrated density of pixels ± SEM) and rather low GFAP levels (1.87 ± 0.18). The exacerbation of EAE (grade 3 of the disease - paraplegia) led to a significant decrease in synaptophysin expression in both untreated (2.77 ± 0.10, P < 0.01) and placebo-treated animals (2.08 ± 0.29, P < 0.01). Nevertheless, the GA treatment resulted in a marked preservation of synaptophysin staining (6.44 ± 0.45, P < 0.01), especially in the motoneuron surroundings, indicating a decreased loss of inputs, this observation being coupled with the fact that GA-treated animals displayed a milder form of the disease (Figure 3, graph I).
The decrease in input elimination after GA treatment occurred in parallel to a significant reduction in astroglial reaction. In this respect, GA treatment reduced GFAP labeling (6.17 ± 0.22) to lower levels than placebo (GFAP: 10.01 ± 0.80, P < 0.01) and untreated EAE-induced specimens (GFAP: 8.24 ± 0.61, P < 0.05). However, the placebo treatment resulted in a statistically significant increase in GFAP labeling compared to untreated EAE-induced rats (P < 0.05). The basal level of GFAP (1.87 ± 0.18) was significantly lower than the levels of the experimental groups (P < 0.01).
Treatment with GA results in a milder form of EAE
The degree of white matter myelination in the spinal cord was determined in Sudan black-stained sections of lumbar spinal cord from normal, placebo and GA-treated animals obtained during the stage of exacerbation of the disease (Figure 3). The degree of myelination was minimal in placebo-treated animals in the paraplegia stage. GA- treated rats presented lower scores and milder clinical signs of the disease and did not reach paraplegia. Also, axonal myelination, although reduced in comparison to normal material, was more preserved than in the placebo group. Representative pictures of normal, EAE-placebo, EAE-GA, and EAE-grade 3 material are shown in Figure 3.
GA treatment preserves the synaptic terminals during the course of EAE
Several synaptic changes were identified at the ultrastructural level during the course of the disease in positive control rats. Chromatolysis, cell nucleus displacement and retraction of synapses from the cell body were observed in alpha motoneurons of the spinal cord. GA treatment reduced these signs, and a larger number of terminals in apposition to the motoneuron cell surface were observed. Representative examples showing the morphology of motoneurons and presynaptic terminals at the dorsal lamina IX can be seen in Figure 4A-C.
In addition to qualitative observations, a detailed analysis of the afferents of these neurons was carried out and the synaptic covering as well as the number and type of boutons were studied. The total covering of the F, S, and C terminals is presented in Figure 4D. A statistically significant preservation of inputs was observed after GA treatment, parallel to a reduction in the maximum score reached after immunization. It is important to emphasize, however, that after treatment the covering remained lower than in the normal situation. A relevant finding is the positive effect of the treatment for both excitatory and inhibitory terminals (Figure 4D-G). In this respect, GA administration preserved the mean length of apposition as well as the number of excitatory and inhibitory inputs (Figure 4E-G).
Pattern of button elimination induced by EAE inflammation
To investigate whether there was a quantitative change in the pattern of synaptic elimination, an analysis of the frequency distribution of terminals apposed to the motoneuron membrane was carried out. A particular feature of the synaptic elimination process is that the retraction of boutons does not occur not by chance, but rather is a finely organized event. In this respect, synaptic removal after lesion tends to preserve clusters of terminals at specific points along the cell membrane of injured neurons. This feature is demonstrated by the fact that, in a normal situation, a number of boutons can be found close together. After injury, however, the distance between groups of terminals tends to increase, an event that can be studied by observing the frequency distribution of the gaps between boutons. Thus, during exacerbation of the disease, the placebo-treated animals displayed an increase in larger intervals, greater than 4 µm, exposing, on average, an additional 35% of the motoneuron membrane. Interestingly, the GA treatment that preserved more terminals in apposition to the motoneurons resulted in a distribution of gaps strikingly similar to that of normal animals (Figure 5).
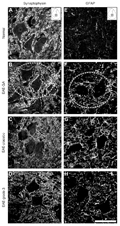 Panels A-D, Immunolabelling against synaptophysin in the normal (A), EAE-GA (B), EAE-placebo (C), and EAE-grade 3 (D) animals. Observe the preservation of inputs in the EAE-GA-treated group (dashed circle in B). Panels E-H, Immunofluorescence micrographs showing astrocyte reactivity in the normal (E), EAE-GA (F), EAE-placebo (G), and EAE-grade 3 (H) animals. Note that a certain degree of astrogliosis is still present in the EAE-GA group, although the clinical signs of the disease have been ameliorated (dashed circle in F). Scale bar = 35 µm for all panels. GFAP = glial fibrillary acidic protein; EAE = experimental autoimmune encephalomyelitis; GA = glatiramer acetate; grade 3 = hind leg paralysis.
Panels A-D, Immunolabelling against synaptophysin in the normal (A), EAE-GA (B), EAE-placebo (C), and EAE-grade 3 (D) animals. Observe the preservation of inputs in the EAE-GA-treated group (dashed circle in B). Panels E-H, Immunofluorescence micrographs showing astrocyte reactivity in the normal (E), EAE-GA (F), EAE-placebo (G), and EAE-grade 3 (H) animals. Note that a certain degree of astrogliosis is still present in the EAE-GA group, although the clinical signs of the disease have been ameliorated (dashed circle in F). Scale bar = 35 µm for all panels. GFAP = glial fibrillary acidic protein; EAE = experimental autoimmune encephalomyelitis; GA = glatiramer acetate; grade 3 = hind leg paralysis.
[View larger version of this image (457 K JPG file)]

Quantitative evaluation of the immunoreactivity under the different experimental conditions, demonstrating the maintenance of synaptic activity after GA treatment. GA treatment significantly decreased the astroglial response in the lumbar spinal cord, when compared to control groups. EAE = experimental autoimmune encephalomyelitis; GA = glatiramer acetate; grade 3 = hind leg paralysis. *P < 0.05 (Student
[View larger version of this image (79 K JPG file)]
 Panels A-D, Sudan black staining at the level of the ventral horn of the spinal cord showing the degree of myelination in normal (A), EAE-GA (peak of the disease after treatment, B), EAE-placebo (C) and EAE- grade 3 (D) animals. Note that GA preserved a significant degree of myelination (dashed circle in B). The placebo-treated and untreated material showed evident signs of demyelination (dashed circles in C and D, respectively). Panels E-H, Toluidine blue-stained semi-thin sections obtained from the different experimental situations. Normal (E), EAE-GA (F), EAE-placebo (G), and EAE-grade 3 (H). Observe the influx of immune cells in the vicinity of the motoneurons (dashed circles in G and H). Scale bar A-D = 35 µm, E-H = 15 µm. The graphs in I and J represent schematic illustrations of the time course of the experimental autoimmune encephalomyelitis and the correlation between the clinical scores after GA and placebo treatment. The peak of the disease occurred at about 14 days after immunization. EAE = experimental autoimmune encephalomyelitis; GA = glatiramer acetate; grade 3 = hind leg paralysis.
Panels A-D, Sudan black staining at the level of the ventral horn of the spinal cord showing the degree of myelination in normal (A), EAE-GA (peak of the disease after treatment, B), EAE-placebo (C) and EAE- grade 3 (D) animals. Note that GA preserved a significant degree of myelination (dashed circle in B). The placebo-treated and untreated material showed evident signs of demyelination (dashed circles in C and D, respectively). Panels E-H, Toluidine blue-stained semi-thin sections obtained from the different experimental situations. Normal (E), EAE-GA (F), EAE-placebo (G), and EAE-grade 3 (H). Observe the influx of immune cells in the vicinity of the motoneurons (dashed circles in G and H). Scale bar A-D = 35 µm, E-H = 15 µm. The graphs in I and J represent schematic illustrations of the time course of the experimental autoimmune encephalomyelitis and the correlation between the clinical scores after GA and placebo treatment. The peak of the disease occurred at about 14 days after immunization. EAE = experimental autoimmune encephalomyelitis; GA = glatiramer acetate; grade 3 = hind leg paralysis.
[View larger version of this image (306 K JPG file)]
 Panel A represents the normal covering of a control rat. Panel B is an example of an EAE-GA specimen - note the normal apposition of the nerve terminal. PanelC, Micrograph of a motoneuron in the placebo-treated situation, showing an almost fully retracted terminal. The colorized images highlight the pre-synaptic terminal in green and the motoneuron cytoplasm and cytoplasmic membrane in gold. Scale bar = 0.5 µm. Panels D-G, Detailed analysis of the synaptic covering and of the number of synaptic boutons after GA treatment, revealing the positive effects of GA compared to the placebo group. Panel D, Graph showing the preservation of both F (flattened vesicles, inhibitory inputs) - and S (spherical vesicles, excitatory inputs) - boutons after GA treatment. Panels E and F, Preservation of both inhibitory and excitatory terminals by GA treatment in both normalized (number of boutons/100 µm of membrane) and non-normalized numbers. Panel G, The button length of the F-, S- and C- (cholinergic) terminals under the different experimental circumstances. EAE = experimental autoimmune encephalomyelitis; GA = glatiramer acetate; grade 3 = hind leg paralysis. *P < 0.05 (Student t-test).
Panel A represents the normal covering of a control rat. Panel B is an example of an EAE-GA specimen - note the normal apposition of the nerve terminal. PanelC, Micrograph of a motoneuron in the placebo-treated situation, showing an almost fully retracted terminal. The colorized images highlight the pre-synaptic terminal in green and the motoneuron cytoplasm and cytoplasmic membrane in gold. Scale bar = 0.5 µm. Panels D-G, Detailed analysis of the synaptic covering and of the number of synaptic boutons after GA treatment, revealing the positive effects of GA compared to the placebo group. Panel D, Graph showing the preservation of both F (flattened vesicles, inhibitory inputs) - and S (spherical vesicles, excitatory inputs) - boutons after GA treatment. Panels E and F, Preservation of both inhibitory and excitatory terminals by GA treatment in both normalized (number of boutons/100 µm of membrane) and non-normalized numbers. Panel G, The button length of the F-, S- and C- (cholinergic) terminals under the different experimental circumstances. EAE = experimental autoimmune encephalomyelitis; GA = glatiramer acetate; grade 3 = hind leg paralysis. *P < 0.05 (Student t-test).
[View larger version of this image (223 K JPG file)]
 Panel A, Normal distribution of intervals between nerve terminals showing that the boutons were organized in clusters of inputs. Panel B, The distribution of terminals during the exacerbation of the disease (grade 3 = hind leg paralysis). Panel C, Treatment with GA keeps the clusters of terminals stable regardless of the increased surrounding glial reaction. Panel D, EAE-placebo, which was similar to the positive control (grade 3 of the disease). EAE = experimental autoimmune encephalomyelitis; GA = glatiramer acetate; F = flattened vesicles, inhibitory inputs; S = spherical vesicles, excitatory inputs; C = cholinergic terminals.
Panel A, Normal distribution of intervals between nerve terminals showing that the boutons were organized in clusters of inputs. Panel B, The distribution of terminals during the exacerbation of the disease (grade 3 = hind leg paralysis). Panel C, Treatment with GA keeps the clusters of terminals stable regardless of the increased surrounding glial reaction. Panel D, EAE-placebo, which was similar to the positive control (grade 3 of the disease). EAE = experimental autoimmune encephalomyelitis; GA = glatiramer acetate; F = flattened vesicles, inhibitory inputs; S = spherical vesicles, excitatory inputs; C = cholinergic terminals.
[View larger version of this image (117 K JPG file)]
The present study supports the view that maintenance of inputs of spinal motoneurons may positively influence their functionality during the course of EAE. This hypothesis is based on the observation that, during the course of the disease, a series of plastic phenomena involving alpha motoneuron afferents correlate with a worsening of the symptoms of the disease (5). Also, a highly plastic response of the motor system after neuroinflammatory lesion was reported by Kerschensteiner et al. (3), with axonal remodeling being observed at multiple levels, including the area surrounding the lesion, and above as well as inside the motor cortex (3).
At present, the treatment of the remittent/recurrent form of multiple sclerosis is based on the administration of glatiramer acetate and beta interferon (4,30,31). Although both are approved drugs for MS treatment, GA has the unique characteristic of being a synthetic polymer composed of L-alanine, L-glutamine, L-lysine and L-tyrosine, exerting a marked suppressive and protective effect on EAE in various animal species (30). Interestingly, although GA is not encephalitogenic, its therapeutic effect is related to the induction of both cellular and humoral immune responses in experimental models and in humans. Currently, its therapeutic effects are attributed to the induction of specific Th2 suppressor cells that, upon an encephalitogenic stimulus, are able to release anti-inflammatory cytokines that minimize the progression of the disease (30). However, apart from such an immunological role, it is possible that GA acts directly on nervous tissue, stimulating the release of trophic and neurotrophic factors by the glial cells, influencing the response to the inflammatory injuries caused by the autoimmune response. In this respect, it has been reported that GA induced an elevation of brain-derived neurotrophic factor, TGF-β, IL-4 and -10 and suppressing TNF-α and INF-γ (10,18,32).
The effects of GA considered above may directly influence the synaptic stability of the spinal cord circuits, which are perturbed during exacerbation of the disease. Analysis of the results obtained by immunohistochemistry in the present study showed that GA treatment significantly preserved synaptophysin labeling in the spinal cord during the period in which the placebo-treated animals presented a great reduction in such immunolabeling. This observation, coupled with the decrease in clinical scores in GA-treated animals, indicates a correlation between synaptic activity and the degree of severity of the disease.
Interestingly, treatment with GA reduced the glial reaction in the surroundings of the alpha motoneurons, as seen by GFAP immunolabeling, as well as at the ultra-structural level. However, the reaction was still more elevated than in the normal situation. This fact strengthens the idea that a certain degree of inflammation was still present in the motor area microenvironment. Another aspect to be considered is that GFAP labeling was greater in placebo-treated animals in comparison to untreated EAE-induced rats. This may indicate that the treatment itself, which consisted of daily subcutaneous injections, may have caused additional stress that influenced the immune response after immunization.
A relevant observation was obtained with Sudan black staining, which revealed the presence of demyelination in GA-treated animals. This indicates that the disease was still progressing, although in a milder way than in the placebo-treated animals. In this respect, it is acceptable to conclude that part of the beneficial effects of treatment is a result of the stabilization of the inputs in the spinal cord microenvironment secondary to the control of the autoimmune response.
The conclusions discussed above were also reinforced by the ultrastructural analysis of the afferents in contact with the alpha motoneurons during the course of the disease. These data indicated that GA treatment decreased the retraction of both excitatory and inhibitory synapses to a level close to normality. Thus, the total synaptic covering seems to have been preserved, apart from the relatively high reactive gliosis, indicating an influence of GA on the synaptic elimination process. The administration of GA also positively influenced the clustering pattern of the terminals, which was preserved, in contrast to the placebo-treated condition (33). Taking into account that a motoneuron receives up to 100,000 inputs from various sources, the maintenance of active synapses is of crucial importance to the correct functionality of the motor pathway.
The present results indicate that the immunomodulator GA influences the plasticity of neural elements in the spinal cord. This may be of relevance in the development of new strategies for the treatment of MS based on the use or combination of drugs capable of acting directly on the synapses, increasing the stability of the neural circuits, which, in turn, may significantly reduce the clinical signs of neurodegenerative diseases.[/body]
 Address for correspondence: A.L.R. Oliveira, Departamento de Anatomia, Instituto de Biologia, UNICAMP, Caixa Postal 6109, 13083-970 Campinas, SP, Brasil. Fax: +55-19-3521-6101. E-mail: alroliv@unicamp.br
Address for correspondence: A.L.R. Oliveira, Departamento de Anatomia, Instituto de Biologia, UNICAMP, Caixa Postal 6109, 13083-970 Campinas, SP, Brasil. Fax: +55-19-3521-6101. E-mail: alroliv@unicamp.br
Research supported by FAPESP (#2004/00498-0). K.B. Marques was supported by a PhD scholarship from CNPq (#304834/2006-8). A.L.R. Oliveira receives a fellowship from CNPq (#304834/2006-8). Publication supported by FAPESP (2008/09949-5). Received June 21, 2008. Accepted January 5, 2009.

Figure 1. Photomicrographs obtained at the level of the spinal cord ventral horn (L4-L5) showing the preservation of synaptophysin and an increase in GFAP and F4/80 during the EAE peak, followed by glatiramer acetate treatment. Panels A-D, Immunolabelling against synaptophysin in the normal (A), EAE-GA (B), EAE-placebo (C), and EAE-grade 3 (D) animals. Observe the preservation of inputs in the EAE-GA-treated group (dashed circle in B). Panels E-H, Immunofluorescence micrographs showing astrocyte reactivity in the normal (E), EAE-GA (F), EAE-placebo (G), and EAE-grade 3 (H) animals. Note that a certain degree of astrogliosis is still present in the EAE-GA group, although the clinical signs of the disease have been ameliorated (dashed circle in F). Scale bar = 35 µm for all panels. GFAP = glial fibrillary acidic protein; EAE = experimental autoimmune encephalomyelitis; GA = glatiramer acetate; grade 3 = hind leg paralysis.

Figure 2. Quantitative evaluation of the immunoreactivity under the different experimental conditions, demonstrating the maintenance of synaptic activity after GA treatment. GA treatment significantly decreased the astroglial response in the lumbar spinal cord, when compared to control groups. EAE = experimental autoimmune encephalomyelitis; GA = glatiramer acetate; grade 3 = hind leg paralysis. *P < 0.05 (Student t-test).

Figure 3. Photomicrographs obtained at the level of the spinal cord ventral horn (L4-L5) showing the correlation between demyelination/inflammation and the clinical scores for EAE. Panels A-D, Sudan black staining at the level of the ventral horn of the spinal cord showing the degree of myelination in normal (A), EAE-GA (peak of the disease after treatment, B), EAE-placebo (C) and EAE- grade 3 (D) animals. Note that GA preserved a significant degree of myelination (dashed circle in B). The placebo-treated and untreated material showed evident signs of demyelination (dashed circles in C and D, respectively). Panels E-H, Toluidine blue-stained semi-thin sections obtained from the different experimental situations. Normal (E), EAE-GA (F), EAE-placebo (G), and EAE-grade 3 (H). Observe the influx of immune cells in the vicinity of the motoneurons (dashed circles in G and H). Scale bar A-D = 35 µm, E-H = 15 µm. The graphs in I and J represent schematic illustrations of the time course of the experimental autoimmune encephalomyelitis and the correlation between the clinical scores after GA and placebo treatment. The peak of the disease occurred at about 14 days after immunization. EAE = experimental autoimmune encephalomyelitis; GA = glatiramer acetate; grade 3 = hind leg paralysis.

Figure 4. Transmission electron microscope micrographs obtained at the level of the spinal cord ventral horn (L4-L5) showing the surface of a motoneuron, where the nerve terminals are in apposition to the plasma membrane (panels A-C). Panel A represents the normal covering of a control rat. Panel B is an example of an EAE-GA specimen - note the normal apposition of the nerve terminal. Panel C, Micrograph of a motoneuron in the placebo-treated situation, showing an almost fully retracted terminal. The colorized images highlight the pre-synaptic terminal in green and the motoneuron cytoplasm and cytoplasmic membrane in gold. Scale bar = 0.5 µm. Panels D-G, Detailed analysis of the synaptic covering and of the number of synaptic boutons after GA treatment, revealing the positive effects of GA compared to the placebo group. Panel D, Graph showing the preservation of both F (flattened vesicles, inhibitory inputs) - and S (spherical vesicles, excitatory inputs) - boutons after GA treatment. Panels E and F, Preservation of both inhibitory and excitatory terminals by GA treatment in both normalized (number of boutons/100 µm of membrane) and non-normalized numbers. Panel G, The button length of the F-, S- and C- (cholinergic) terminals under the different experimental circumstances. EAE = experimental autoimmune encephalomyelitis; GA = glatiramer acetate; grade 3 = hind leg paralysis. *P < 0.05 (Student t-test).

Figure 5. Transmission electron microscope micrographs obtained at the level of the spinal cord ventral horn (L4-L5) showing that the distribution of the gap length between clusters of terminals apposing the cell soma membrane was close to normality after GA treatment. Panel A, Normal distribution of intervals between nerve terminals showing that the boutons were organized in clusters of inputs. Panel B, The distribution of terminals during the exacerbation of the disease (grade 3 = hind leg paralysis). Panel C, Treatment with GA keeps the clusters of terminals stable regardless of the increased surrounding glial reaction. Panel D, EAE-placebo, which was similar to the positive control (grade 3 of the disease). EAE = experimental autoimmune encephalomyelitis; GA = glatiramer acetate; F = flattened vesicles, inhibitory inputs; S = spherical vesicles, excitatory inputs; C = cholinergic terminals.
- 1. Trapp BD, Bo L, Mork S, Chang A. Pathogenesis of tissue injury in MS lesions. J Neuroimmunol 1999; 98: 49-56.
- 2. Kornek B, Storch MK, Weissert R, Wallstroem E, Stefferl A, Olsson T, et al. Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am J Pathol 2000; 157: 267-276.
- 3. Kerschensteiner M, Bareyre FM, Buddeberg BS, Merkler D, Stadelmann C, Bruck W, et al. Remodeling of axonal connections contributes to recovery in an animal model of multiple sclerosis. J Exp Med 2004; 200: 1027-1038.
- 4. Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain 2006; 129: 1953-1971.
- 5. Marques KB, Santos LM, Oliveira AL. Spinal motoneuron synaptic plasticity during the course of an animal model of multiple sclerosis. Eur J Neurosci 2006; 24: 3053-3062.
- 6. Lassmann H, Bruck W, Lucchinetti C. Heterogeneity of multiple sclerosis pathogenesis: implications for diagnosis and therapy. Trends Mol Med 2001; 7: 115-121.
- 7. Hemmer B, Archelos JJ, Hartung HP. New concepts in the immunopathogenesis of multiple sclerosis. Nat Rev Neurosci 2002; 3: 291-301.
- 8. Cross AH, Stark JL. Humoral immunity in multiple sclerosis and its animal model, experimental autoimmune encephalomyelitis. Immunol Res 2005; 32: 85-97.
- 9. Schmidt CS, Zhao J, Chain J, Hepburn D, Gitter B, Sandusky G, et al. Resistance to myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis by death receptor 6-deficient mice. J Immunol 2005; 175: 2286-2292.
- 10. Ziemssen T, Ziemssen F. The role of the humoral immune system in multiple sclerosis (MS) and its animal model experimental autoimmune encephalomyelitis (EAE). Autoimmun Rev 2005; 4: 460-467.
- 11. Wegner C. Pathological differences in acute inflammatory demyelinating diseases of the central nervous system. Int MS J 2005; 12: 13-19.
- 12. Merkler D, Boscke R, Schmelting B, Czeh B, Fuchs E, Bruck W, et al. Differential macrophage/microglia activation in neocortical EAE lesions in the marmoset monkey. Brain Pathol 2006; 16: 117-123.
- 13. Kust B, Mantingh-Otter I, Boddeke E, Copray S. Deficient p75 low-affinity neurotrophin receptor expression does alter the composition of cellular infiltrate in experimental autoimmune encephalomyelitis in C57BL/6 mice. J Neuroimmunol 2006; 174: 92-100.
- 14. Li XB, Schluesener HJ. Therapeutic effects of cisplatin on rat experimental autoimmune encephalomyelitis. Arch Immunol Ther Exp 2006; 54: 51-53.
- 15. Liu Y, Liu J, Tetzlaff W, Paty DW, Cynader MS. Biliverdin reductase, a major physiologic cytoprotectant, suppresses experimental autoimmune encephalomyelitis. Free Radic Biol Med 2006; 40: 960-967.
- 16. Muthian G, Raikwar HP, Rajasingh J, Bright JJ. 1,25 Dihydroxyvitamin-D3 modulates JAK-STAT pathway in IL-12/IFNgamma axis leading to Th1 response in experimental allergic encephalomyelitis. J Neurosci Res 2006; 83: 1299-1309.
- 17. Vogler S, Pahnke J, Rousset S, Ricquier D, Moch H, Miroux B, et al. Uncoupling protein 2 has protective function during experimental autoimmune encephalomyelitis. Am J Pathol 2006; 168: 1570-1575.
- 18. Aharoni R, Kayhan B, Eilam R, Sela M, Arnon R. Glatiramer acetate-specific T cells in the brain express T helper 2/3 cytokines and brain-derived neurotrophic factor in situ Proc Natl Acad Sci U S A 2003; 100: 14157-14162.
- 19. Gilgun-Sherki Y, Panet H, Holdengreber V, Mosberg-Galili R, Offen D. Axonal damage is reduced following glatiramer acetate treatment in C57/bl mice with chronic-induced experimental autoimmune encephalomyelitis. Neurosci Res 2003; 47: 201-207.
- 20. Racke MK, Martin R, McFarland H, Fritz RB. Copolymer-1-induced inhibition of antigen-specific T cell activation: interference with antigen presentation. J Neuroimmunol 1992; 37: 75-84.
- 21. Teitelbaum D, Milo R, Arnon R, Sela M. Synthetic copolymer 1 inhibits human T-cell lines specific for myelin basic protein. Proc Natl Acad Sci U S A 1992; 89: 137-141.
- 22. Fridkis-Hareli M, Teitelbaum D, Gurevich E, Pecht I, Brautbar C, Kwon OJ, et al. Direct binding of myelin basic protein and synthetic copolymer 1 to class II major histocompatibility complex molecules on living antigen-presenting cells - specificity and promiscuity. Proc Natl Acad Sci U S A 1994; 91: 4872-4876.
- 23. Teitelbaum D, Sela M, Arnon R. Copolymer 1 from the laboratory to FDA. Isr J Med Sci 1997; 33: 280-284.
- 24. Kipnis J, Yoles E, Porat Z, Cohen A, Mor F, Sela M, et al. T cell immunity to copolymer 1 confers neuroprotection on the damaged optic nerve: possible therapy for optic neuropathies. Proc Natl Acad Sci U S A 2000; 97: 7446-7451.
- 25. Schori H, Kipnis J, Yoles E, WoldeMussie E, Ruiz G, Wheeler LA, et al. Vaccination for protection of retinal ganglion cells against death from glutamate cytotoxicity and ocular hypertension: implications for glaucoma. Proc Natl Acad Sci U S A 2001; 98: 3398-3403.
- 26. Angelov DN, Waibel S, Guntinas-Lichius O, Lenzen M, Neiss WF, Tomov TL, et al. Therapeutic vaccine for acute and chronic motor neuron diseases: implications for amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 2003; 100: 4790-4795.
- 27. Ziemssen T, Kumpfel T, Klinkert WE, Neuhaus O, Hohlfeld R. Glatiramer acetate-specific T-helper 1- and 2-type cell lines produce BDNF: implications for multiple sclerosis therapy. Brain-derived neurotrophic factor. Brain 2002; 125: 2381-2391.
- 28. Emirandetti A, Graciele ZR, Sabha M Jr, De Oliveira AL. Astrocyte reactivity influences the number of presynaptic terminals apposed to spinal motoneurons after axotomy. Brain Res 2006; 1095: 35-42.
- 29. Conradi S, Skoglund S. Observations on the ultrastructure of the initial motor axon segment and dorsal root boutons on the motoneurons in the lumbosacral spinal cord of the cat during postnatal development. Acta Physiol Scand Suppl 1969; 333: 53-76.
- 30. Dhib-Jalbut S. Glatiramer acetate (Copaxone) therapy for multiple sclerosis. Pharmacol Ther 2003; 98: 245-255.
- 31. Polman CH, Uitdehaag BM. New and emerging treatment options for multiple sclerosis. Lancet Neurol 2003; 2: 563-566.
- 32. Hammarberg H, Lidman O, Lundberg C, Eltayeb SY, Gielen AW, Muhallab S, et al. Neuroprotection by encephalomyelitis: rescue of mechanically injured neurons and neurotrophin production by CNS-infiltrating T and natural killer cells. J Neurosci 2000; 20: 5283-5291.
- 33. Cullheim S, Wallquist W, Hammarberg H, Linda H, Piehl F, Carlstedt T, et al. Properties of motoneurons underlying their regenerative capacity after axon lesions in the ventral funiculus or at the surface of the spinal cord. Brain Res Brain Res Rev 2002; 40: 309-316.
Correspondence and Footnotes

Publication Dates
-
Publication in this collection
09 Mar 2009 -
Date of issue
Feb 2009
History
-
Received
21 June 2008 -
Accepted
05 Jan 2009
